

USH2A-retinopathy: From genetics to therapeutics
- PMID: 33121974
- PMCID: PMC8417766
- DOI: 10.1016/j.exer.2020.108330
USH2A-retinopathy: From genetics to therapeutics
Abstract
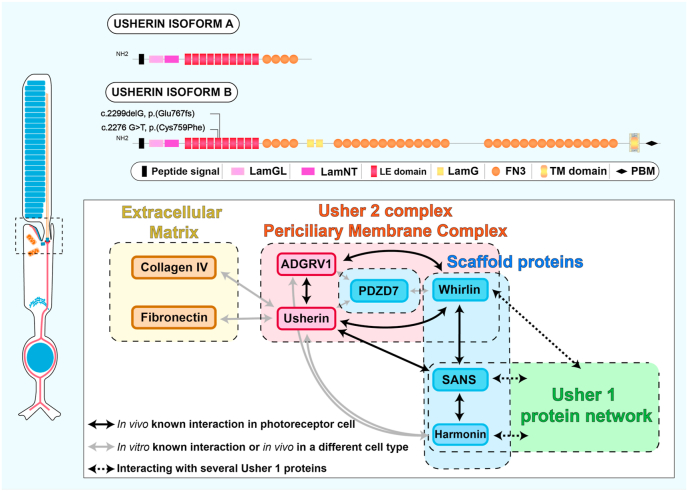
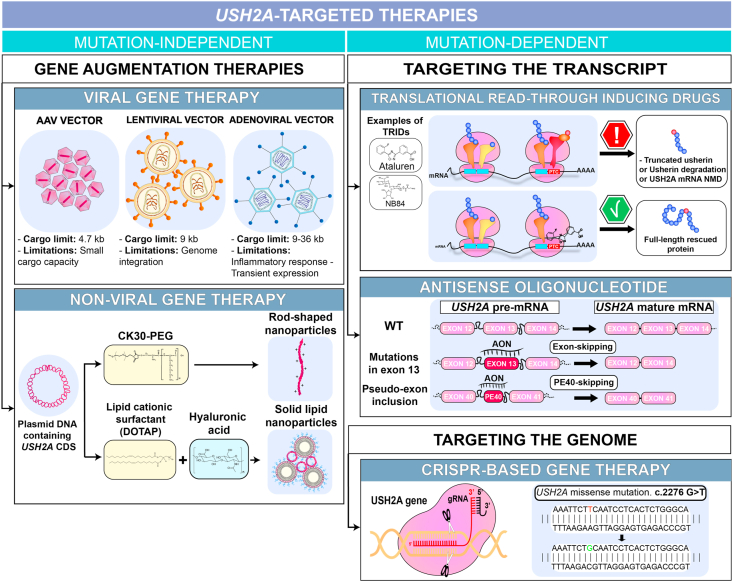
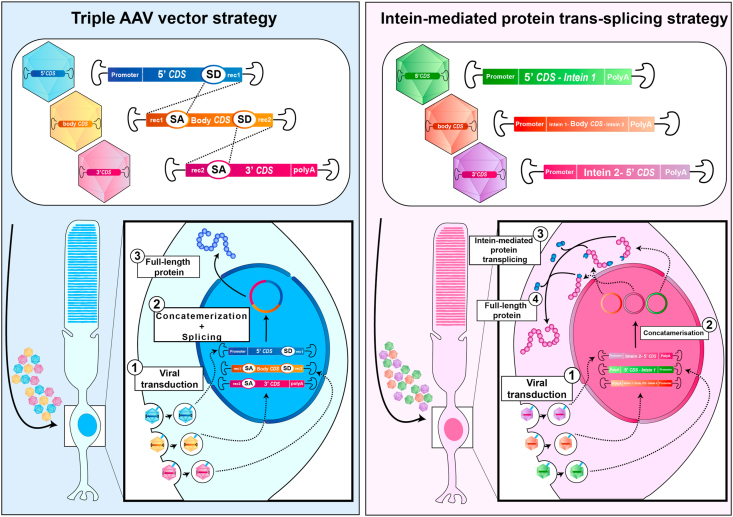
Bilallelic variants in the USH2A gene can cause Usher syndrome type 2 and non-syndromic retinitis pigmentosa. In both disorders, the retinal phenotype involves progressive rod photoreceptor loss resulting in nyctalopia and a constricted visual field, followed by subsequent cone degeneration, leading to the loss of central vision and severe visual impairment. The USH2A gene raises many challenges for researchers and clinicians due to a broad spectrum of mutations, a large gene size hampering gene therapy development and limited knowledge on its pathogenicity. Patients with Usher type 2 may benefit from hearing aids or cochlear implants to correct their hearing defects, but there are currently no approved treatments available for the USH2A-retinopathy. Several treatment strategies, including antisense oligonucleotides and translational readthrough inducing drugs, have shown therapeutic promise in preclinical studies. Further understanding of the pathogenesis and natural history of USH2A-related disorders is required to develop innovative treatments and design clinical trials based on reliable outcome measures. The present review will discuss the current knowledge about USH2A, the emerging therapeutics and existing challenges.
Keywords: Disease models; Hair cells; Photoreceptor; Retinitis pigmentosa; Therapy; USH2A; Usher syndrome; Usherin.
Copyright © 2020 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Adato A., Lefèvre G., Delprat B., Michel V., Michalski N., Chardenoux S., Weil D., El-Amraoui A., Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. - DOI - PubMed
-
- Ahmed Z.M., Jaworek T.J., Sarangdhar G.N., Zheng L., Gul K., Khan S.N., Friedman T.B., Sisk R.A., Bartles J.R., Riazuddin Sheikh, Riazuddin Saima. Inframe deletion of human ESPN is associated with deafness, vestibulopathy and vision impairment. J. Med. Genet. 2018;55:479–488. doi: 10.1136/jmedgenet-2017-105221. - DOI - PMC - PubMed
-
- Aller E., Larrieu L., Jaijo T., Baux D., Espinós C., González-Candelas F., Nájera C., Palau F., Claustres M., Roux A.-F., Millán J.M. The USH2A c.2299delG mutation: dating its common origin in a Southern European population. Eur. J. Hum. Genet. 2010;18:788–793. doi: 10.1038/ejhg.2010.14. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
