

Functional in vivo and in vitro effects of 20q11.21 genetic aberrations on hPSC differentiation
- PMID: 33122739
- PMCID: PMC7596514
- DOI: 10.1038/s41598-020-75657-7
Functional in vivo and in vitro effects of 20q11.21 genetic aberrations on hPSC differentiation
Abstract
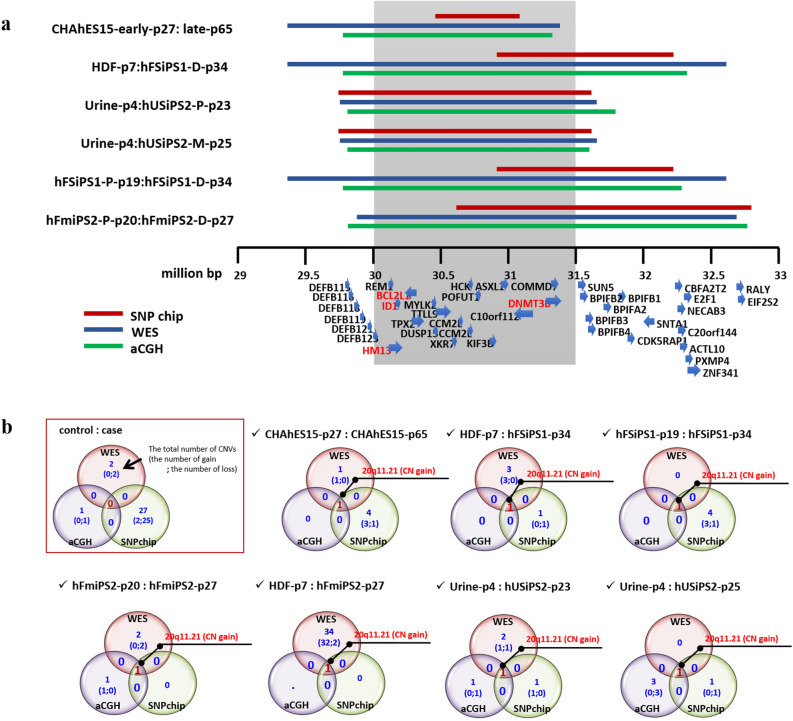
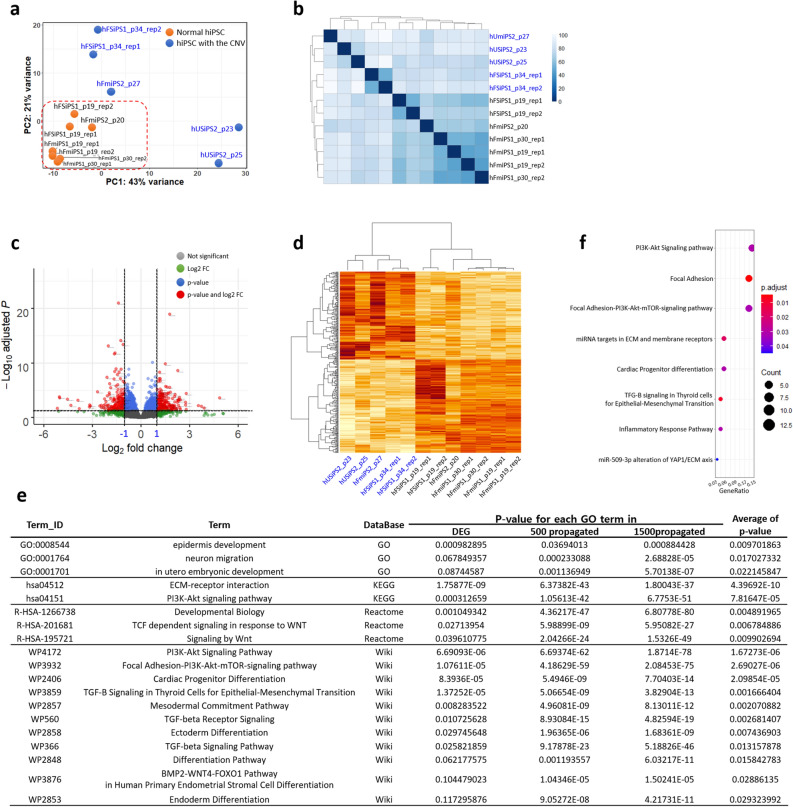
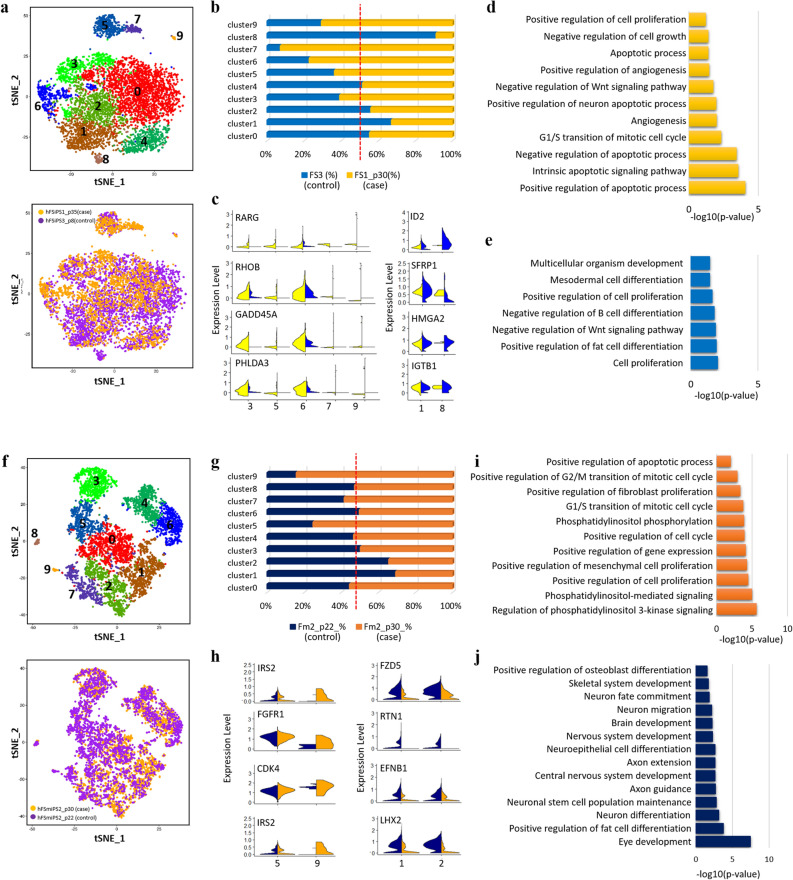
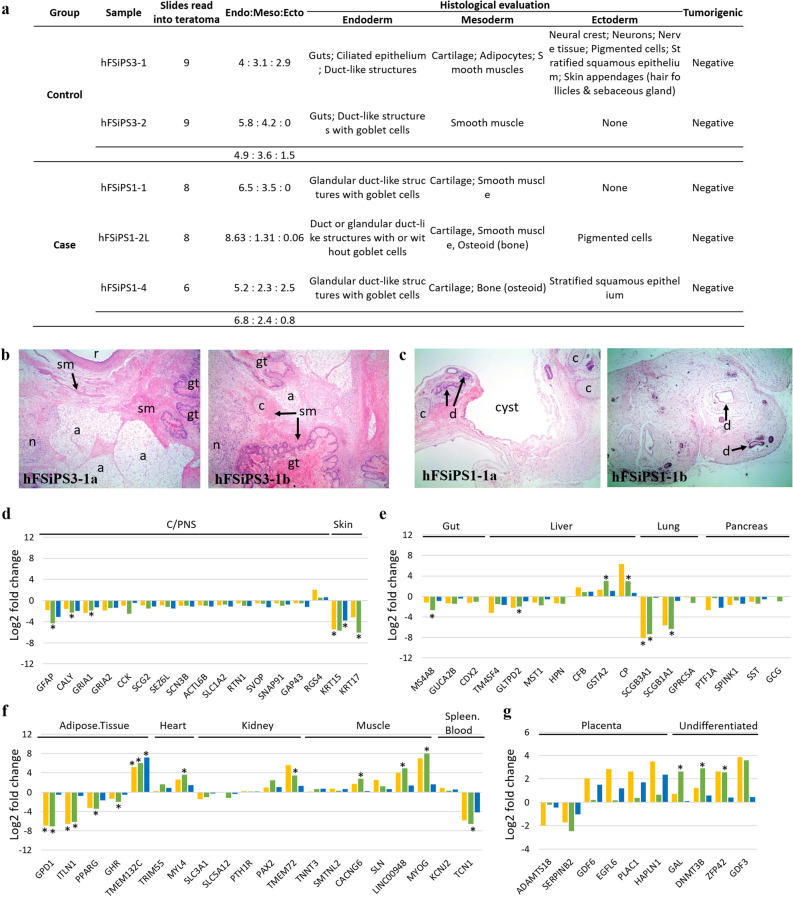
Human pluripotent stem cells (hPSCs) have promising therapeutic applications due to their infinite capacity for self-renewal and pluripotency. Genomic stability is imperative for the clinical use of hPSCs; however, copy number variation (CNV), especially recurrent CNV at 20q11.21, may contribute genomic instability of hPSCs. Furthermore, the effects of CNVs in hPSCs at the whole-transcriptome scale are poorly understood. This study aimed to examine the functional in vivo and in vitro effects of frequently detected CNVs at 20q11.21 during early-stage differentiation of hPSCs. Comprehensive transcriptome profiling of abnormal hPSCs revealed that the differential gene expression patterns had a negative effect on differentiation potential. Transcriptional heterogeneity identified by single-cell RNA sequencing (scRNA-seq) of embryoid bodies from two different isogenic lines of hPSCs revealed alterations in differentiated cell distributions compared with that of normal cells. RNA-seq analysis of 22 teratomas identified several differentially expressed lineage-specific markers in hPSCs with CNVs, consistent with the histological results of the altered ecto/meso/endodermal ratio due to CNVs. Our results suggest that CNV amplification contributes to cell proliferation, apoptosis, and cell fate specification. This work shows the functional consequences of recurrent genetic abnormalities and thereby provides evidence to support the development of cell-based applications.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
