

Empirical comparison of analytical approaches for identifying molecular HIV-1 clusters
- PMID: 33122765
- PMCID: PMC7596705
- DOI: 10.1038/s41598-020-75560-1
Empirical comparison of analytical approaches for identifying molecular HIV-1 clusters
Abstract
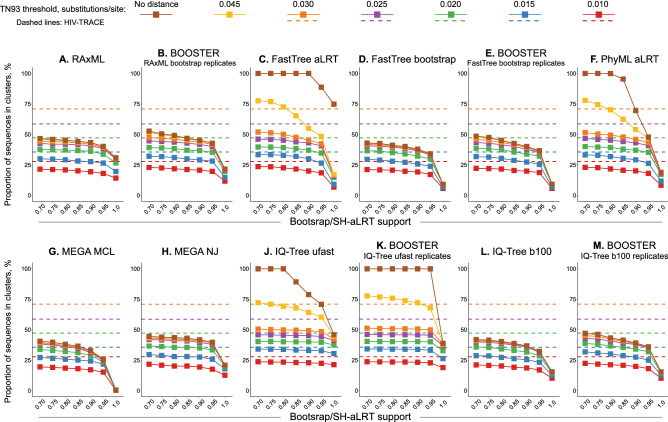
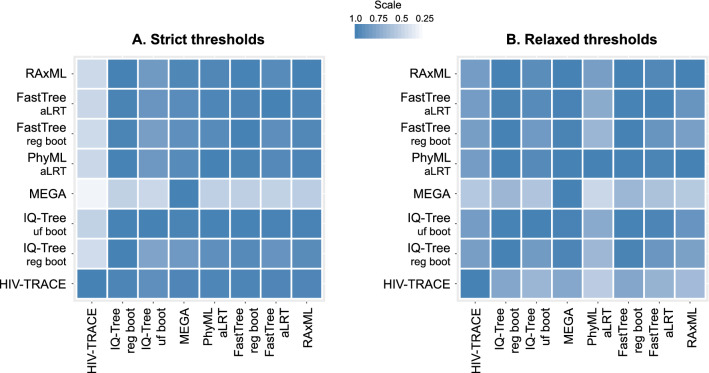
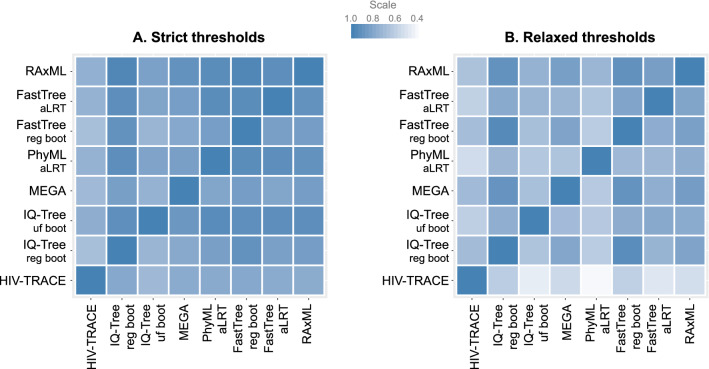
Public health interventions guided by clustering of HIV-1 molecular sequences may be impacted by choices of analytical approaches. We identified commonly-used clustering analytical approaches, applied them to 1886 HIV-1 Rhode Island sequences from 2004-2018, and compared concordance in identifying molecular HIV-1 clusters within and between approaches. We used strict (topological support ≥ 0.95; distance 0.015 substitutions/site) and relaxed (topological support 0.80-0.95; distance 0.030-0.045 substitutions/site) thresholds to reflect different epidemiological scenarios. We found that clustering differed by method and threshold and depended more on distance than topological support thresholds. Clustering concordance analyses demonstrated some differences across analytical approaches, with RAxML having the highest (91%) mean summary percent concordance when strict thresholds were applied, and three (RAxML-, FastTree regular bootstrap- and IQ-Tree regular bootstrap-based) analytical approaches having the highest (86%) mean summary percent concordance when relaxed thresholds were applied. We conclude that different analytical approaches can yield diverse HIV-1 clustering outcomes and may need to be differentially used in diverse public health scenarios. Recognizing the variability and limitations of commonly-used methods in cluster identification is important for guiding clustering-triggered interventions to disrupt new transmissions and end the HIV epidemic.
Conflict of interest statement
MH reports fees from Competition Economics and The Miriam Hospital for consulting, outside the submitted work. All other authors declare that they have no competing interests.
Figures



References
-
- UNAIDS. Global HIV and AIDS statistics—2019 fact sheet. https://www.unaids.org/en/resources/fact-sheet (2019).
-
- 2CDC. Detecting and responding to HIV transmission clusters. A guide for health departments. https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18... (2018).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
