

Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients With Variable Disease Severities
- PMID: 33123498
- PMCID: PMC7572854
- DOI: 10.3389/fcimb.2020.575613
Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients With Variable Disease Severities
Abstract
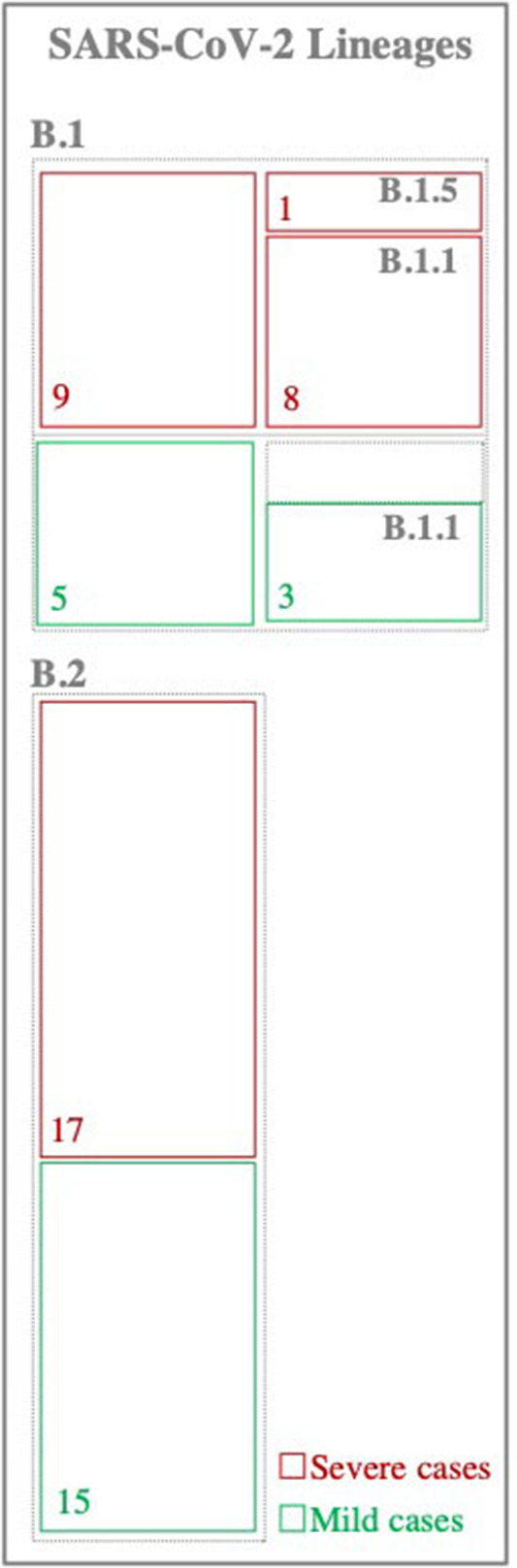
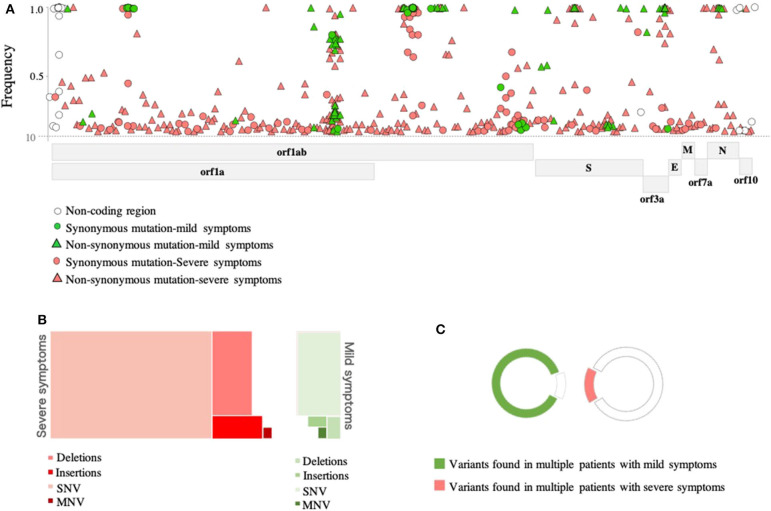
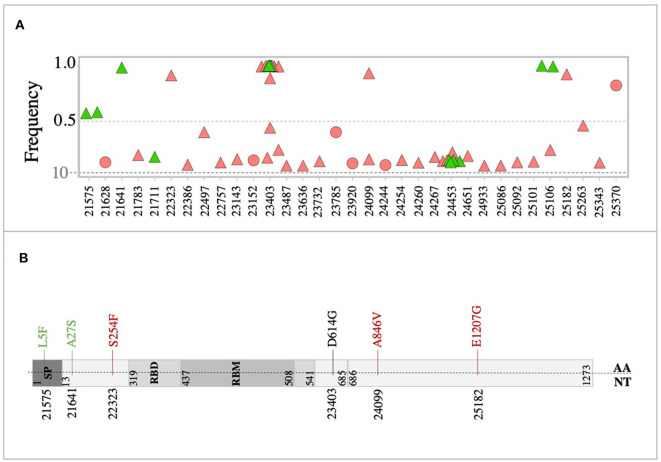
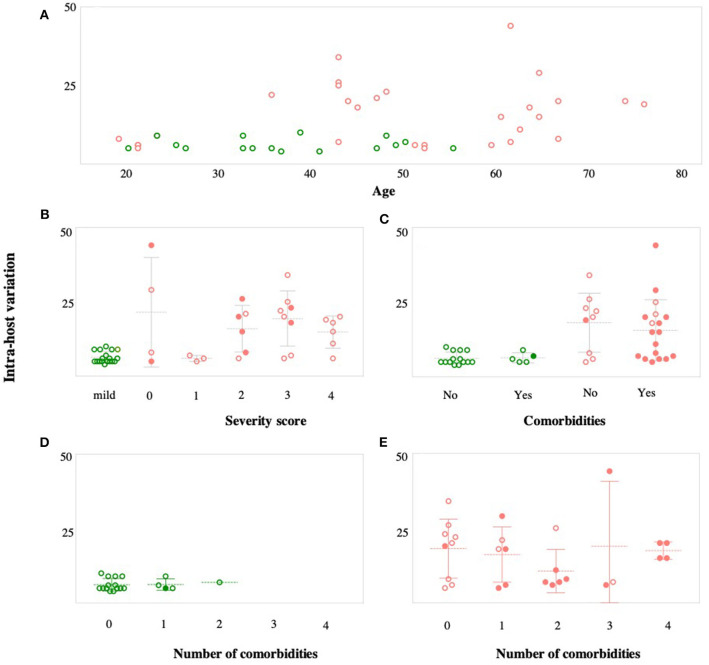
Background: The ongoing pandemic of SARS-COV-2 has already infected more than eight million people worldwide. The majority of COVID-19 patients either are asymptomatic or have mild symptoms. Yet, about 15% of the cases experience severe complications and require intensive care. Factors determining disease severity are not yet fully characterized. Aim: Here, we investigated the within-host virus diversity in COVID-19 patients with different clinical manifestations. Methods: We compared SARS-COV-2 genetic diversity in 19 mild and 27 severe cases. Viral RNA was extracted from nasopharyngeal samples and sequenced using the Illumina MiSeq platform. This was followed by deep-sequencing analyses of SARS-CoV-2 genomes at both consensus and sub-consensus sequence levels. Results: Consensus sequences of all viruses were very similar, showing more than 99.8% sequence identity regardless of the disease severity. However, the sub-consensus analysis revealed significant differences in within-host diversity between mild and severe cases. Patients with severe symptoms exhibited a significantly (p-value 0.001) higher number of variants in coding and non-coding regions compared to mild cases. Analysis also revealed higher prevalence of some variants among severe cases. Most importantly, severe cases exhibited significantly higher within-host diversity (mean = 13) compared to mild cases (mean = 6). Further, higher within-host diversity was observed in patients above the age of 60 compared to the younger age group. Conclusion: These observations provided evidence that within-host diversity might play a role in the development of severe disease outcomes in COVID-19 patients; however, further investigations are required to elucidate this association.
Keywords: COVID-19 severity; SARS-CoV-2; nonsynonymous mutations; virus quasispecies; within-host diversity.
Copyright © 2020 Al Khatib, Benslimane, Elbashir, Coyle, Al Maslamani, Al-Khal, Al Thani and Yassine.
Figures



References
-
- Briese T., Mishra N., Jain K., Zalmout I. S., Jabado O. J., Karesh W. B., et al. (2014). Middle East respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. MBio 5:e01146–14. 10.1128/mBio.01146-14 - DOI - PMC - PubMed
-
- Cabot B., Martell M., Esteban J. I., Piron M., Otero T., Esteban R., et al. (2001). Longitudinal evaluation of the structure of replicating and circulating hepatitis C virus quasispecies in nonprogressive chronic hepatitis C patients. J. Virol. 75, 12005–12013. 10.1128/JVI.75.24.12005-12013.2001 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous