

Machine learning in chemical reaction space
- PMID: 33127879
- PMCID: PMC7603480
- DOI: 10.1038/s41467-020-19267-x
Machine learning in chemical reaction space
Abstract
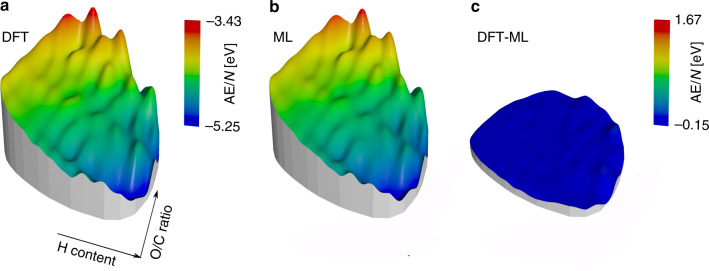
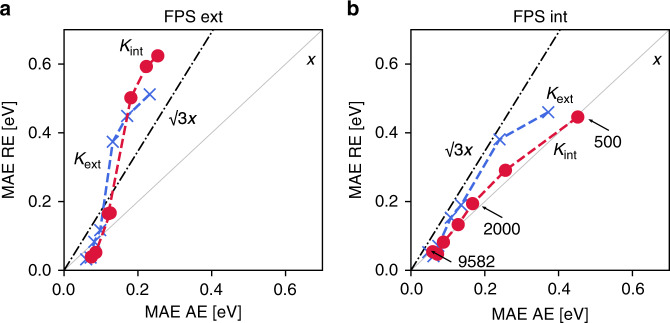
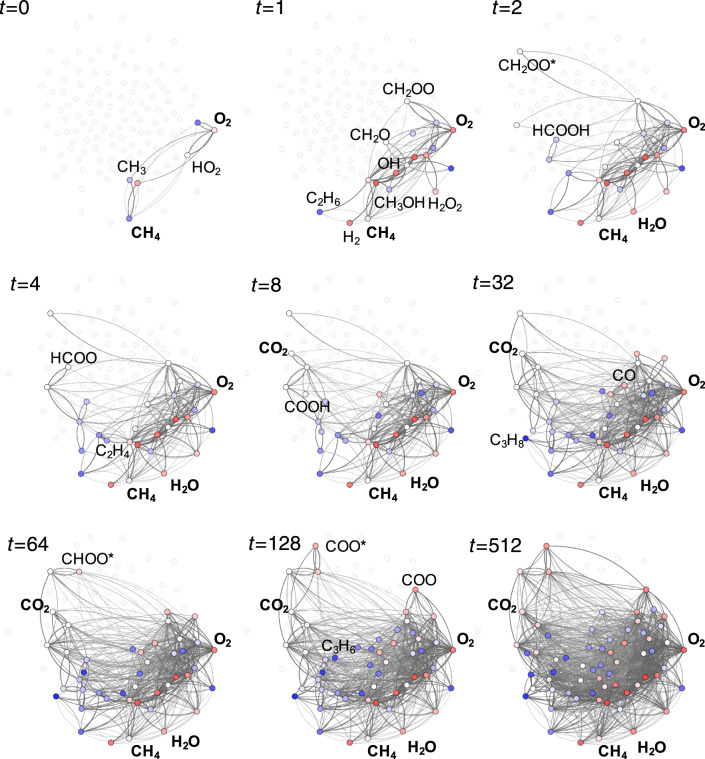
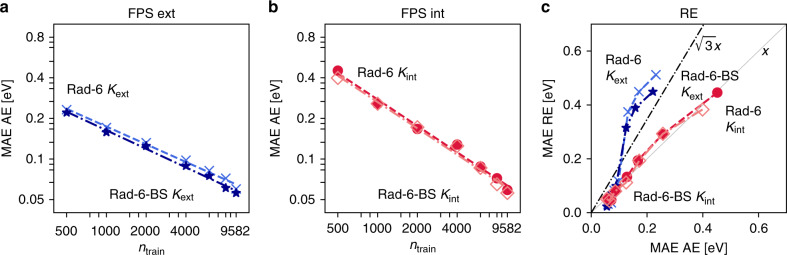
Chemical compound space refers to the vast set of all possible chemical compounds, estimated to contain 1060 molecules. While intractable as a whole, modern machine learning (ML) is increasingly capable of accurately predicting molecular properties in important subsets. Here, we therefore engage in the ML-driven study of even larger reaction space. Central to chemistry as a science of transformations, this space contains all possible chemical reactions. As an important basis for 'reactive' ML, we establish a first-principles database (Rad-6) containing closed and open-shell organic molecules, along with an associated database of chemical reaction energies (Rad-6-RE). We show that the special topology of reaction spaces, with central hub molecules involved in multiple reactions, requires a modification of existing compound space ML-concepts. Showcased by the application to methane combustion, we demonstrate that the learned reaction energies offer a non-empirical route to rationally extract reduced reaction networks for detailed microkinetic analyses.
Conflict of interest statement
The authors declare no competing interests.
Figures
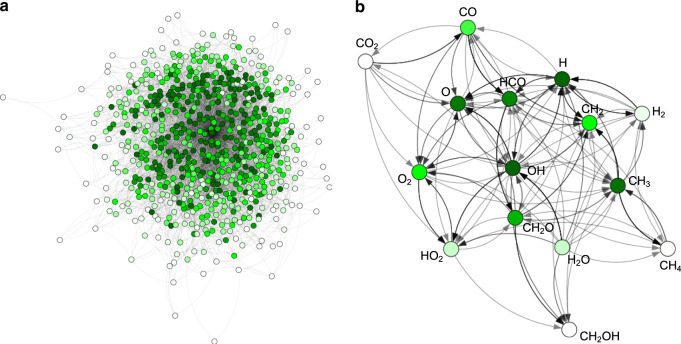
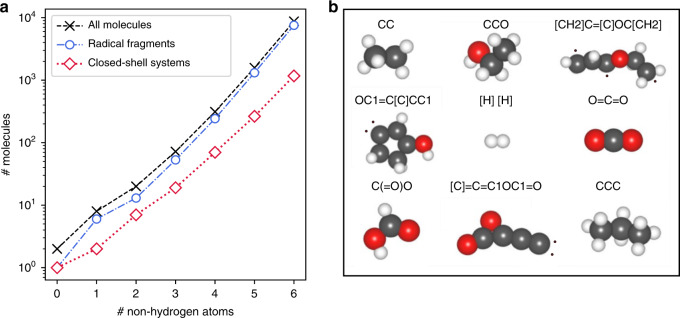
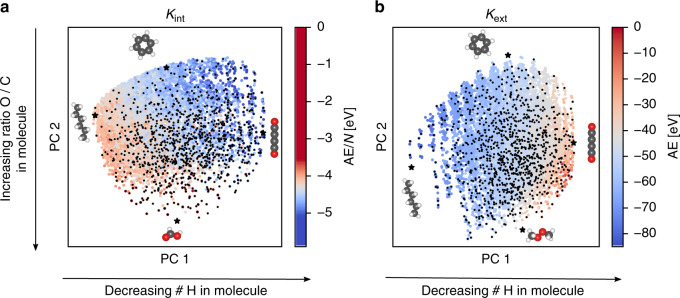
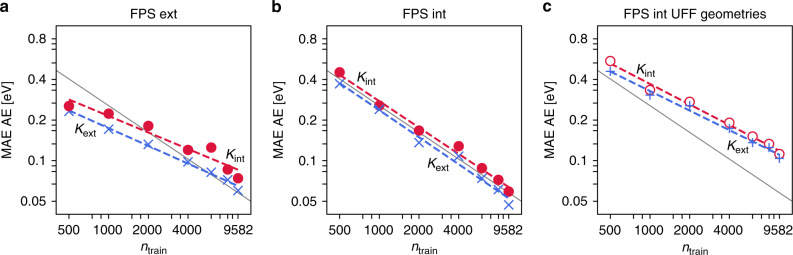
References
-
- Gossler H, Maier L, Angeli S, Tischer S, Deutschmann O. CaRMeN: an improved computer-aided method for developing catalytic reaction mechanisms. Catalysts. 2019;9:227. doi: 10.3390/catal9030227. - DOI
-
- Zhu H, Kee RJ, Janardhanan VM, Deutschmann O, Goodwin DG. Modeling elementary heterogeneous chemistry and electrochemistry in solid-oxide fuel cells. J. Electrochem. Soc. 2005;152:A2427. doi: 10.1149/1.2116607. - DOI
-
- Deutschmann O, Schmidt LD. Modeling the partial oxidation of methane in a short-contact-time reactor. AIChE J. 1998;44:2465–2477. doi: 10.1002/aic.690441114. - DOI
-
- Harper MR, Geem KMV, Pyl SP, Marin GB, Green WH. Comprehensive reaction mechanism for n-butanol pyrolysis and combustion. Combust. Flame. 2011;158:16–41. doi: 10.1016/j.combustflame.2010.06.002. - DOI
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
