

Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity
- PMID: 33128048
- PMCID: PMC8135089
- DOI: 10.1038/s41588-020-00726-6
Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity
Abstract
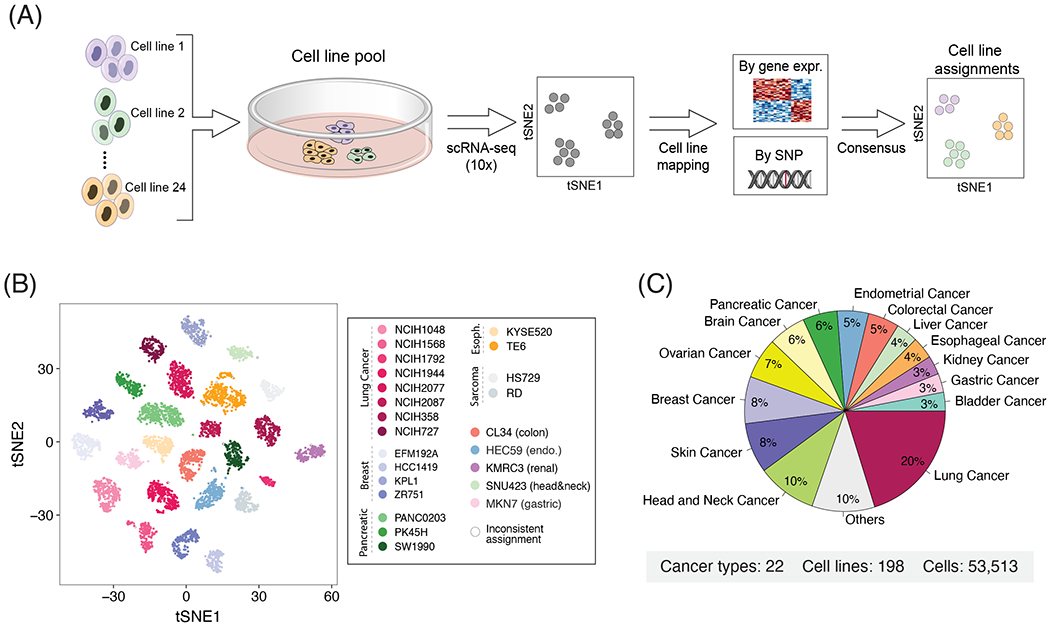
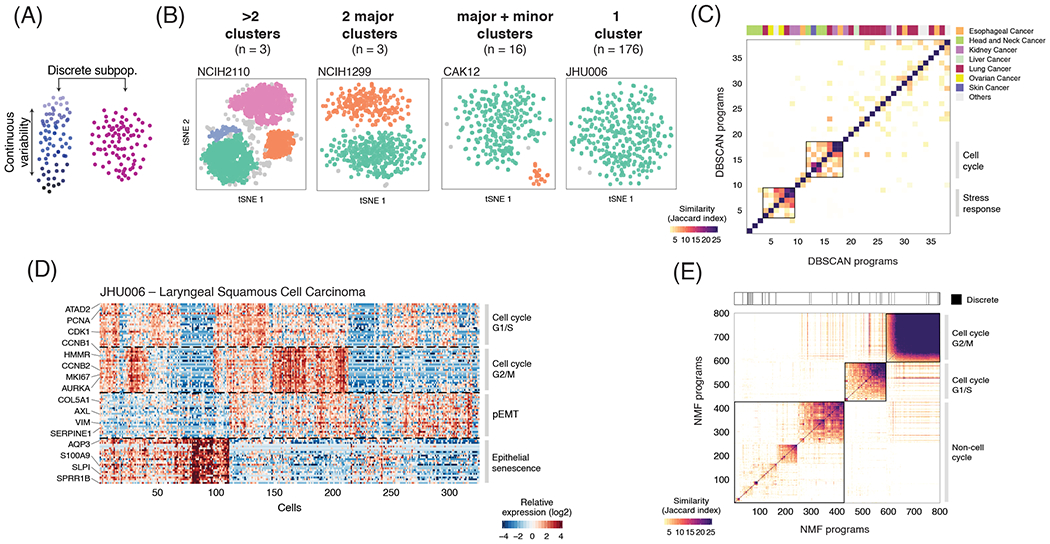
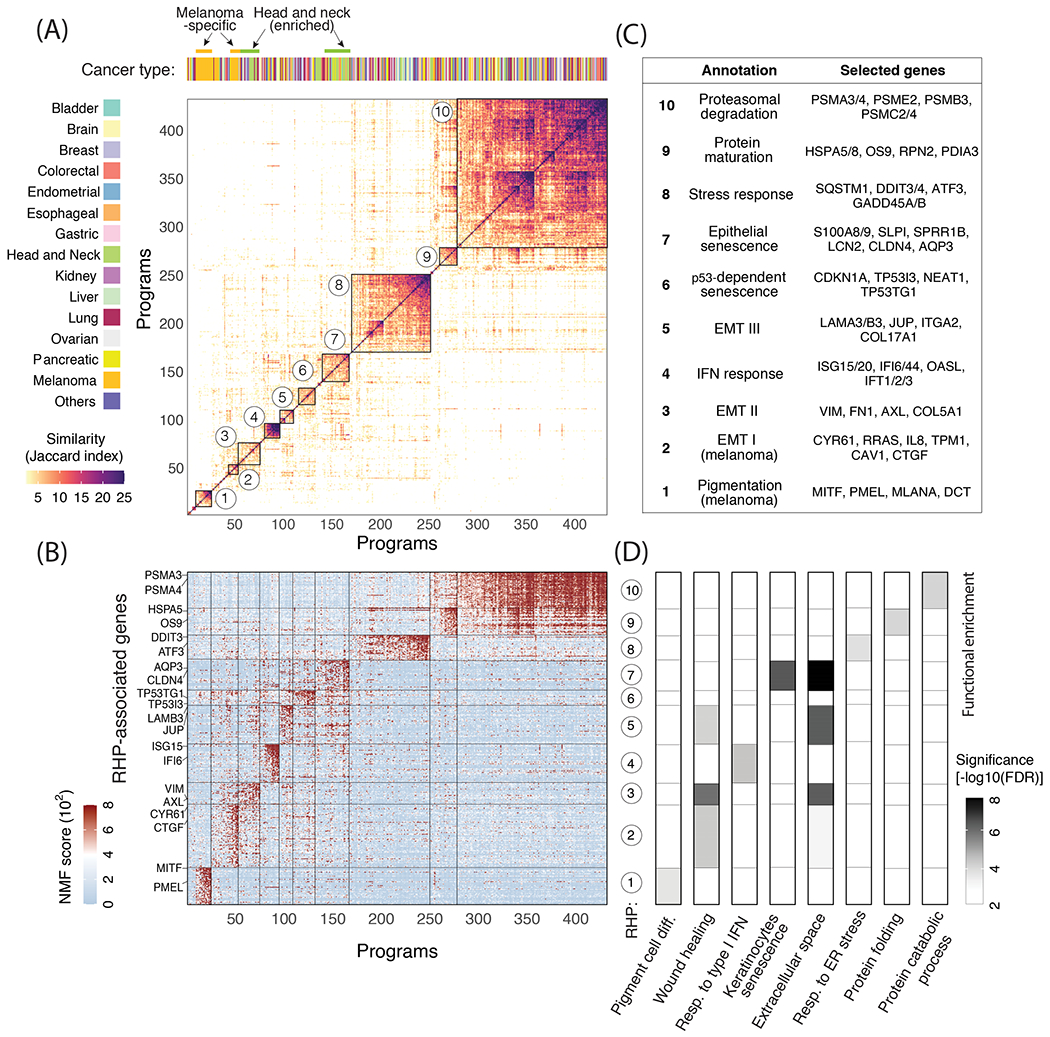
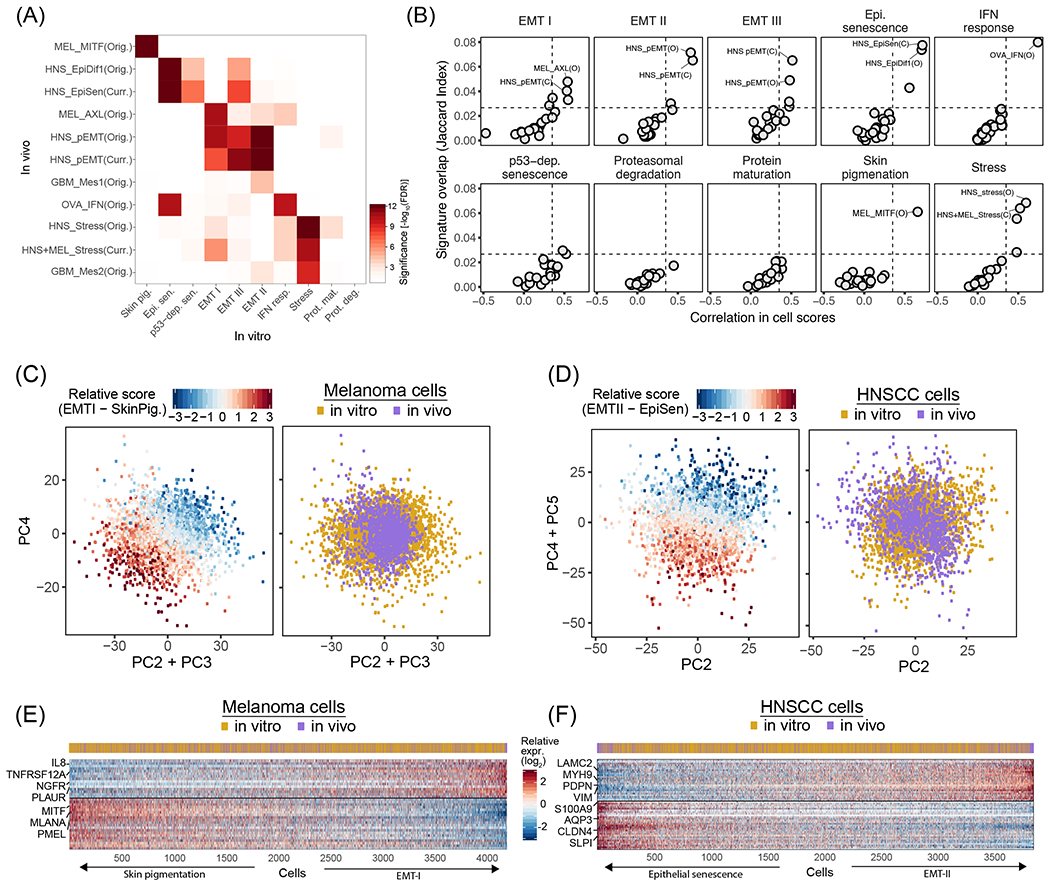
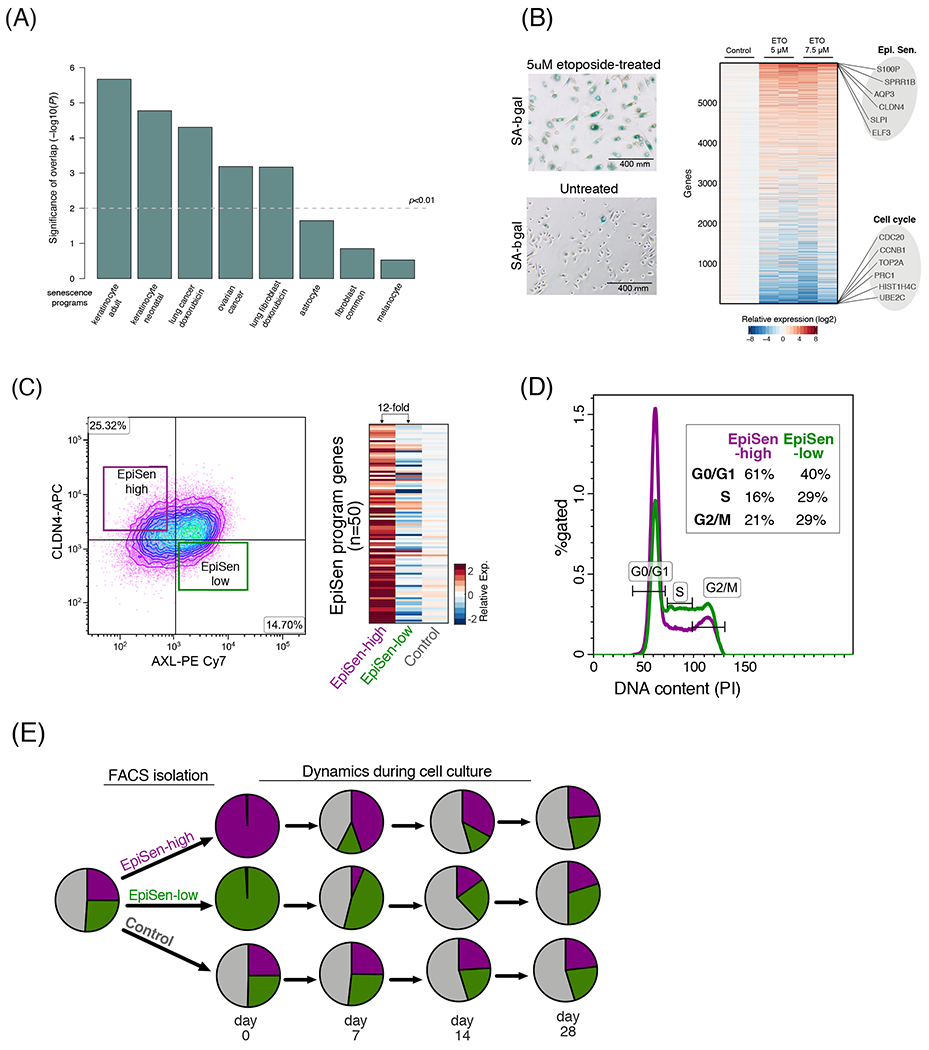
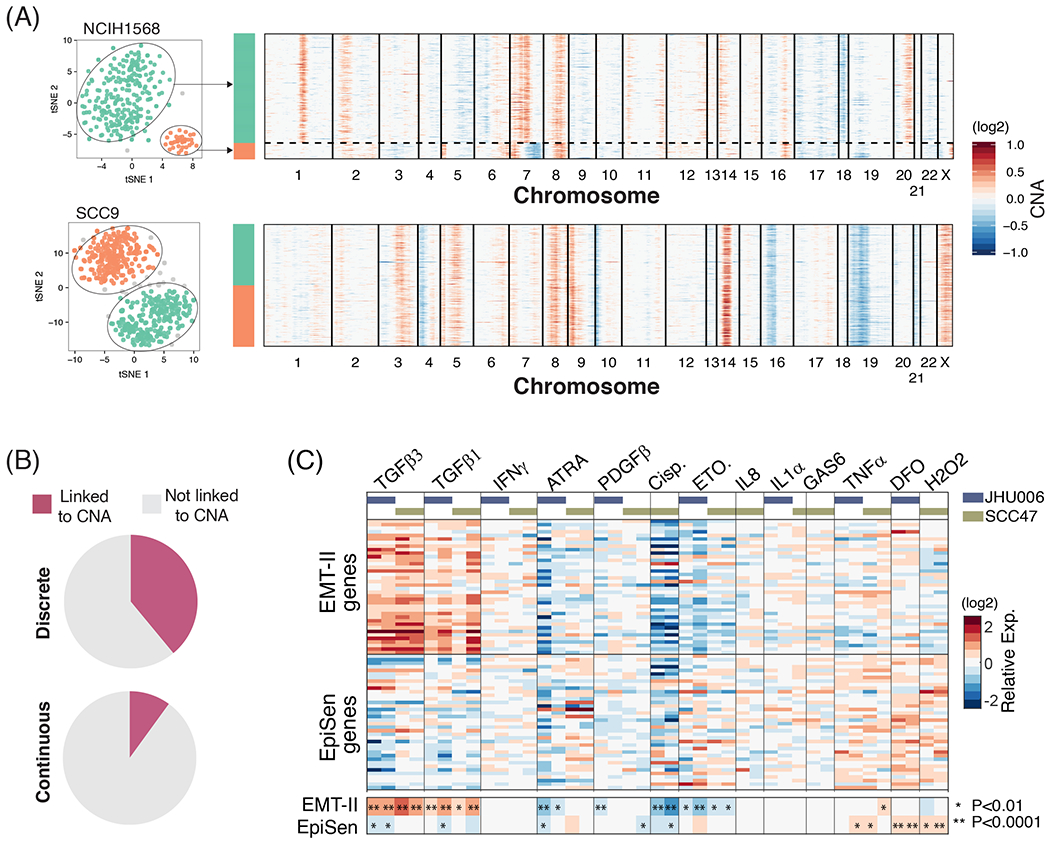
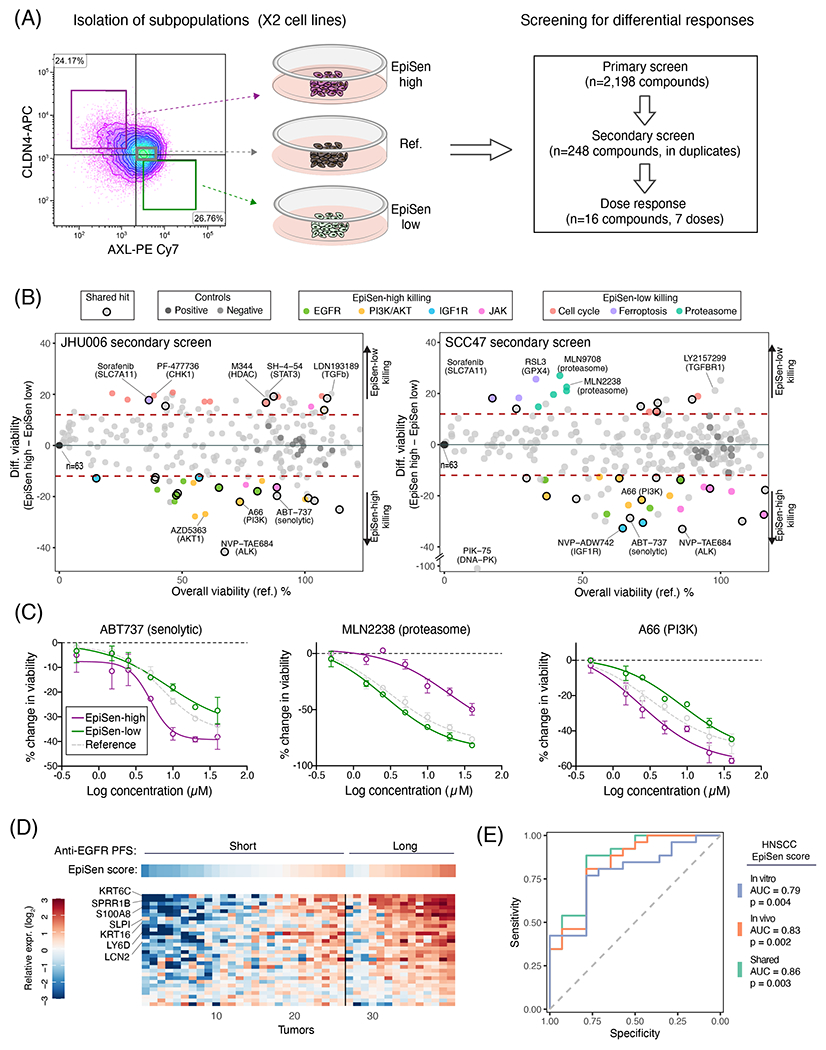
Cultured cell lines are the workhorse of cancer research, but the extent to which they recapitulate the heterogeneity observed among malignant cells in tumors is unclear. Here we used multiplexed single-cell RNA-seq to profile 198 cancer cell lines from 22 cancer types. We identified 12 expression programs that are recurrently heterogeneous within multiple cancer cell lines. These programs are associated with diverse biological processes, including cell cycle, senescence, stress and interferon responses, epithelial-mesenchymal transition and protein metabolism. Most of these programs recapitulate those recently identified as heterogeneous within human tumors. We prioritized specific cell lines as models of cellular heterogeneity and used them to study subpopulations of senescence-related cells, demonstrating their dynamics, regulation and unique drug sensitivities, which were predictive of clinical response. Our work describes the landscape of heterogeneity within diverse cancer cell lines and identifies recurrent patterns of heterogeneity that are shared between tumors and specific cell lines.
Conflict of interest statement
Competing interests
A.R. is a founder and equity holder of Celsius Therapeutics and a SAB member of Neogene Therapeutics, ThermoFisher Scientific and Syros Pharmaceuticals.
Figures
Comment in
-
Cell Lines Recapitulate Intratumoral Heterogeneity Observed In Vivo.Cancer Discov. 2021 Jan;11(1):11. doi: 10.1158/2159-8290.CD-RW2020-166. Epub 2020 Nov 13. Cancer Discov. 2021. PMID: 33188054
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
