

TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions
- PMID: 33130440
- PMCID: PMC7600390
- DOI: 10.1016/j.redox.2020.101755
TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions
Abstract
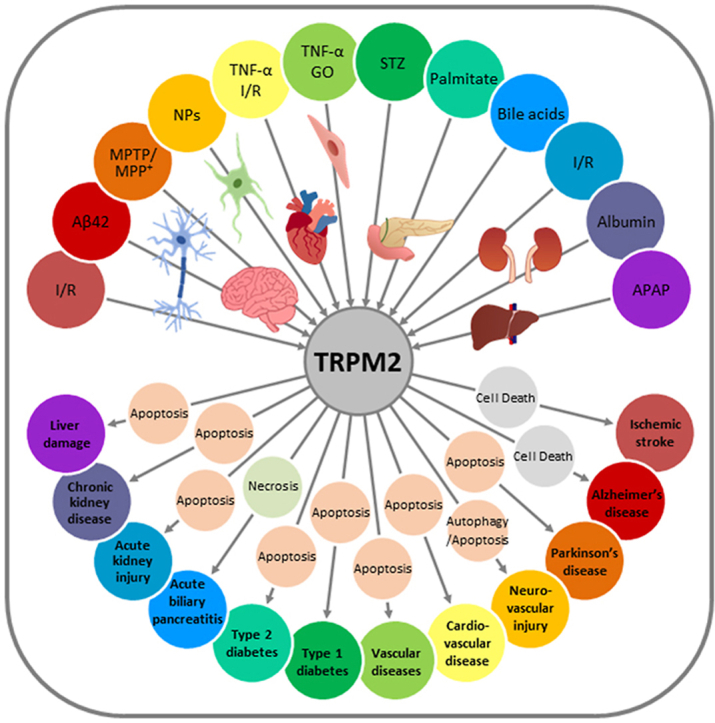
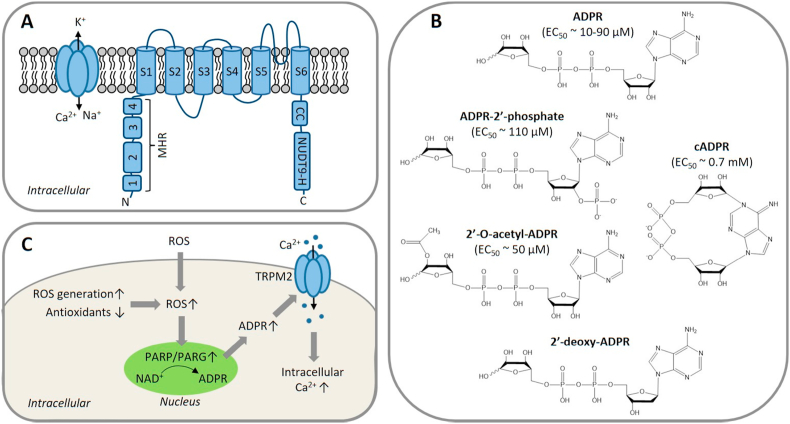
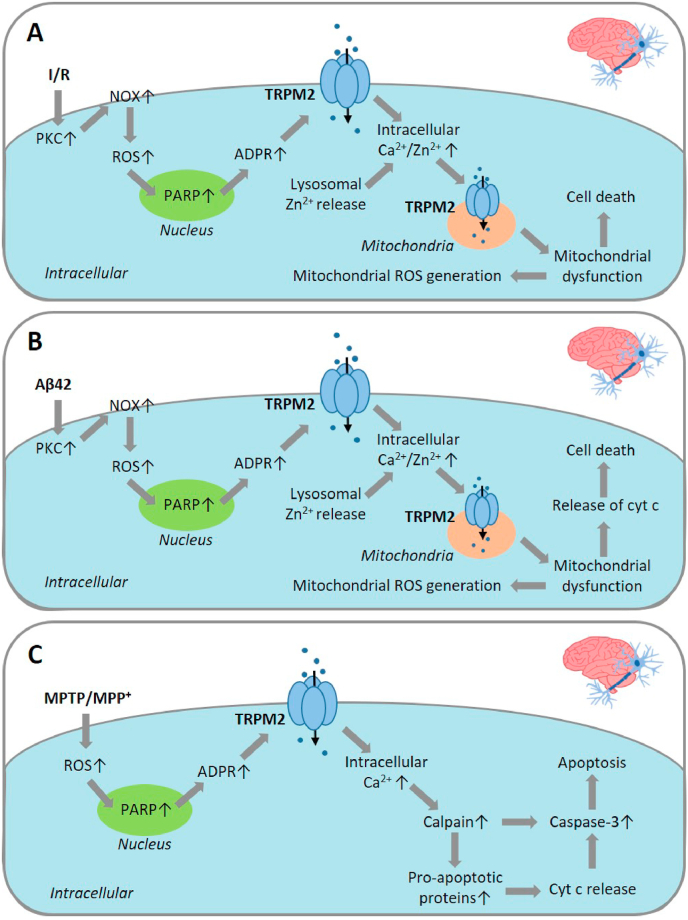
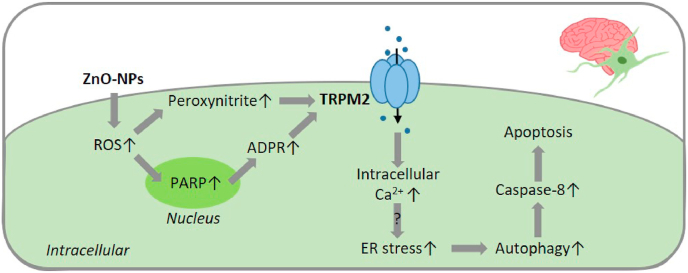
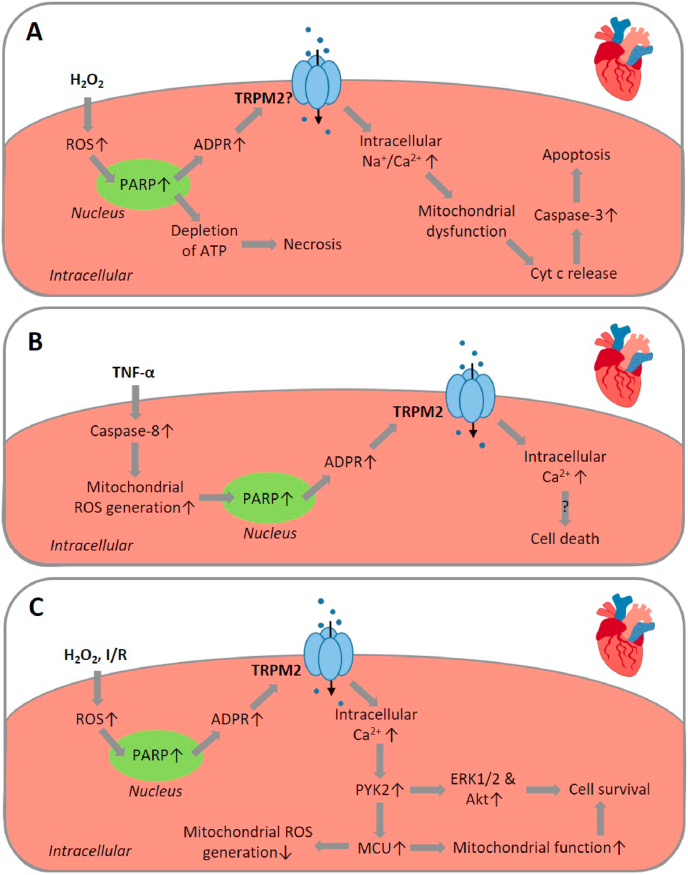
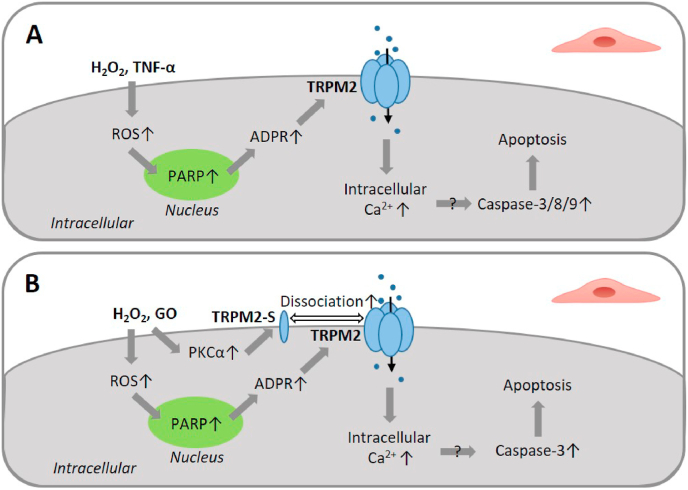
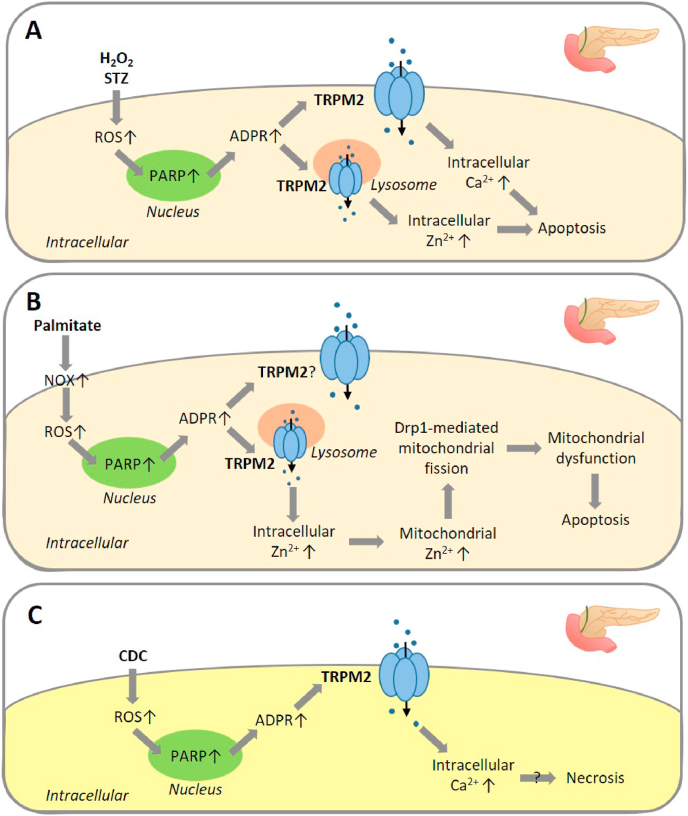
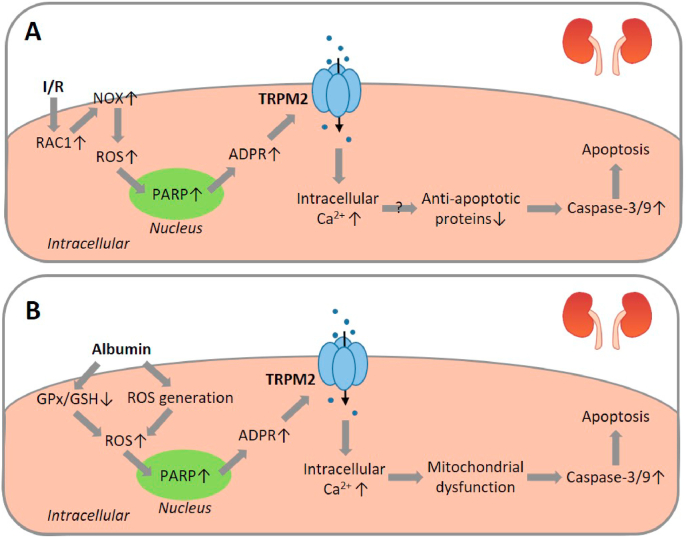
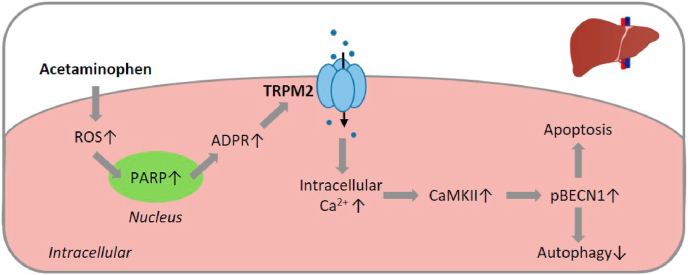
Oxidative stress resulting from the accumulation of high levels of reactive oxygen species is a salient feature of, and a well-recognised pathological factor for, diverse pathologies. One common mechanism for oxidative stress damage is via the disruption of intracellular ion homeostasis to induce cell death. TRPM2 is a non-selective Ca2+-permeable cation channel with a wide distribution throughout the body and is highly sensitive to activation by oxidative stress. Recent studies have collected abundant evidence to show its important role in mediating cell death induced by miscellaneous oxidative stress-inducing pathological factors, both endogenous and exogenous, including ischemia/reperfusion and the neurotoxicants amyloid-β peptides and MPTP/MPP+ that cause neuronal demise in the brain, myocardial ischemia/reperfusion, proinflammatory mediators that disrupt endothelial function, diabetogenic agent streptozotocin and diabetes risk factor free fatty acids that induce loss of pancreatic β-cells, bile acids that damage pancreatic acinar cells, renal ischemia/reperfusion and albuminuria that are detrimental to kidney cells, acetaminophen that triggers hepatocyte death, and nanoparticles that injure pericytes. Studies have also shed light on the signalling mechanisms by which these pathological factors activate the TRPM2 channel to alter intracellular ion homeostasis leading to aberrant initiation of various cell death pathways. TRPM2-mediated cell death thus emerges as an important mechanism in the pathogenesis of conditions including ischemic stroke, neurodegenerative diseases, cardiovascular diseases, diabetes, pancreatitis, chronic kidney disease, liver damage and neurovascular injury. These findings raise the exciting perspective of targeting the TRPM2 channel as a novel therapeutic strategy to treat such oxidative stress-associated diseases.
Keywords: Ca(2+) and Zn(2+) homeostasis; Cell death; Diseases; Oxidative stress; TRPM2 channel.
Copyright © 2020 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
We declare no conflict of interest.
Figures
References
-
- Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
