

Identification of a Metabolic, Transcriptomic, and Molecular Signature of Patatin-Like Phospholipase Domain Containing 3-Mediated Acceleration of Steatohepatitis
- PMID: 33131062
- PMCID: PMC8046714
- DOI: 10.1002/hep.31609
Identification of a Metabolic, Transcriptomic, and Molecular Signature of Patatin-Like Phospholipase Domain Containing 3-Mediated Acceleration of Steatohepatitis
Abstract
Background and aims: The mechanisms by which the I148M mutant variant of the patatin-like phospholipase domain-containing 3 (PNPLA3I148M ) drives development of nonalcoholic steatohepatitis (NASH) are not known. The aim of this study was to obtain insights on mechanisms underlying PNPLA3I148M -induced acceleration of NASH.
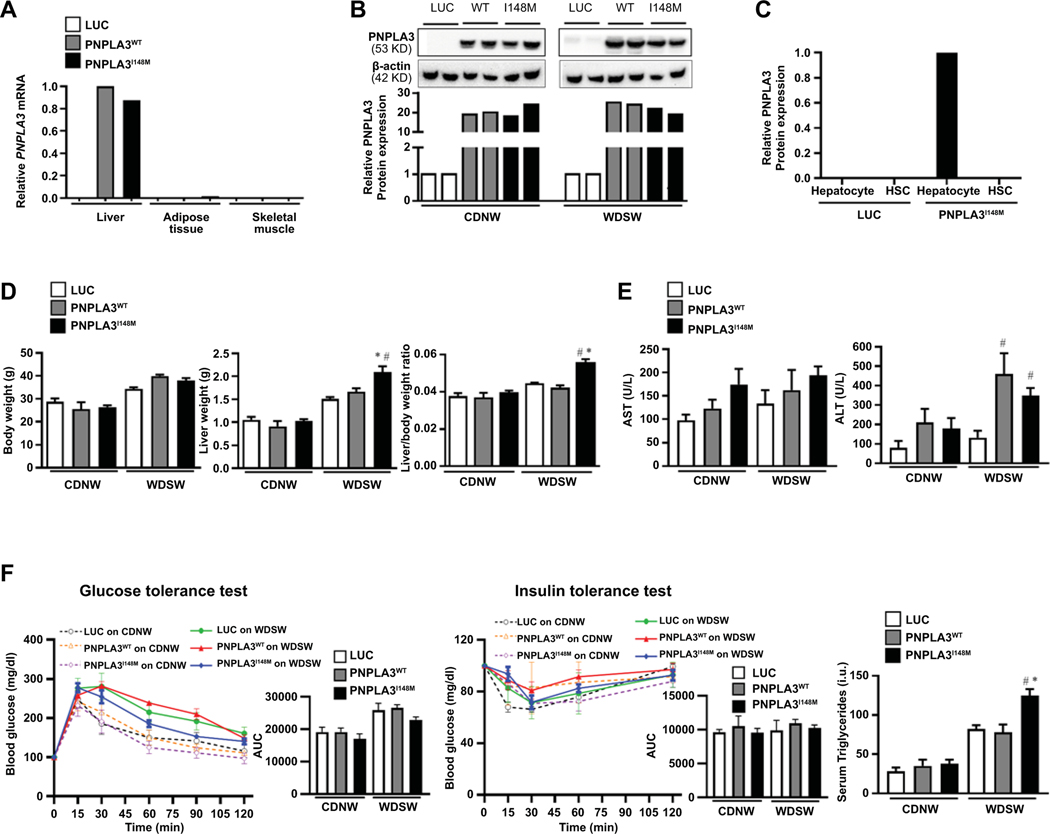
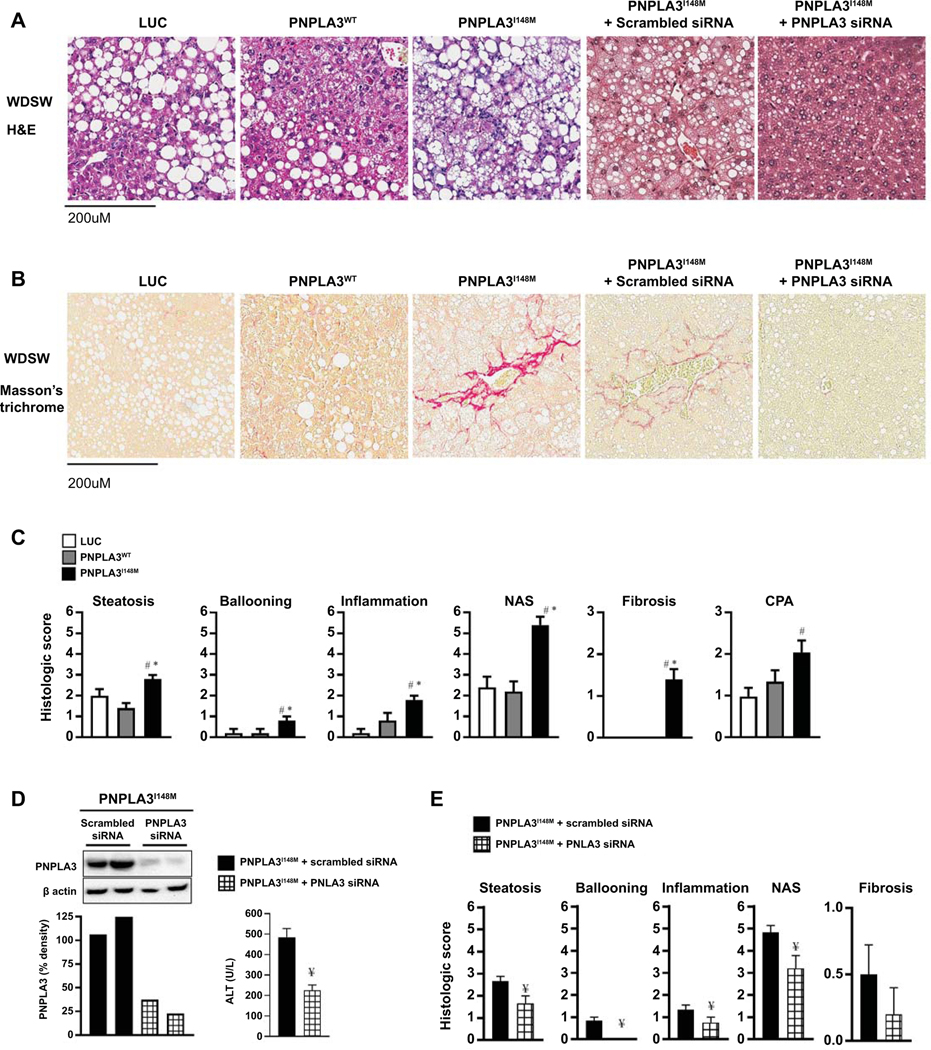
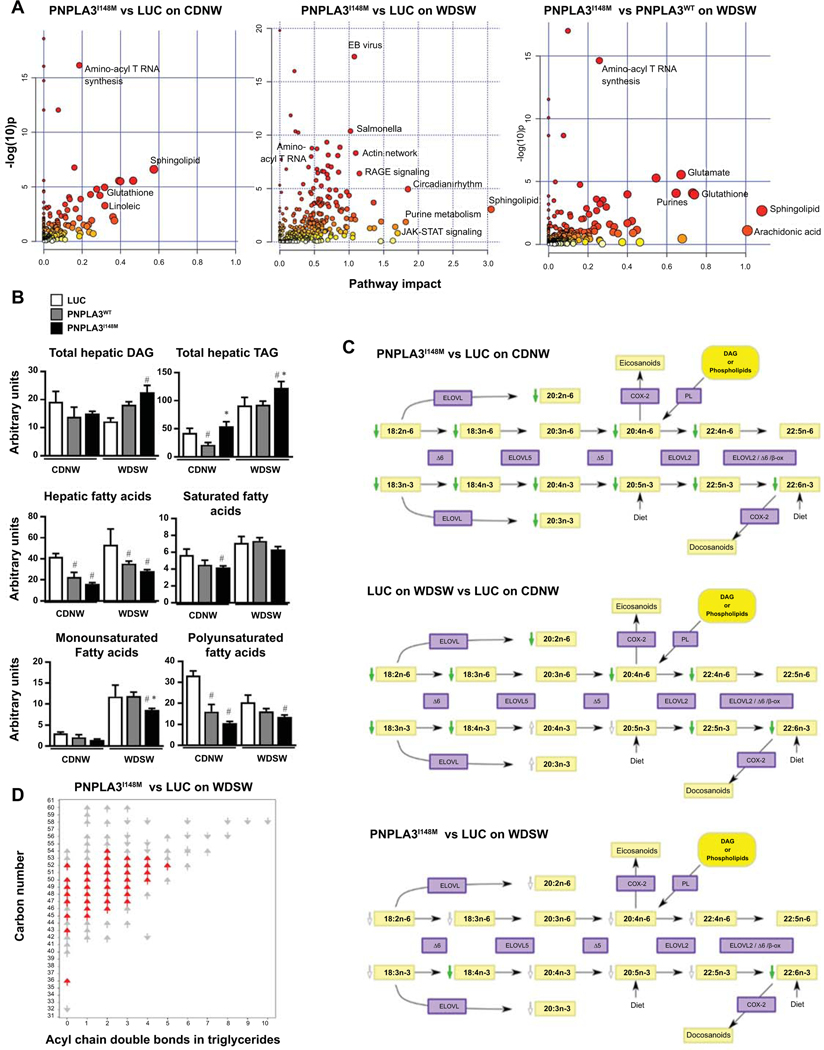
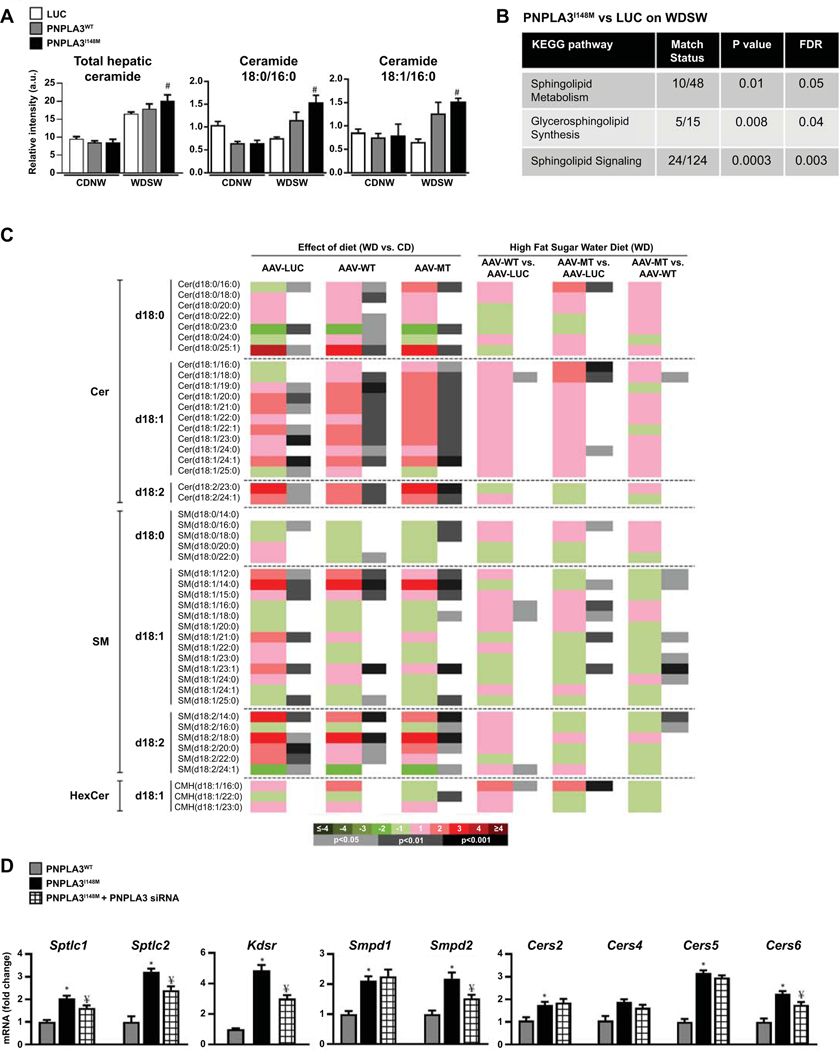
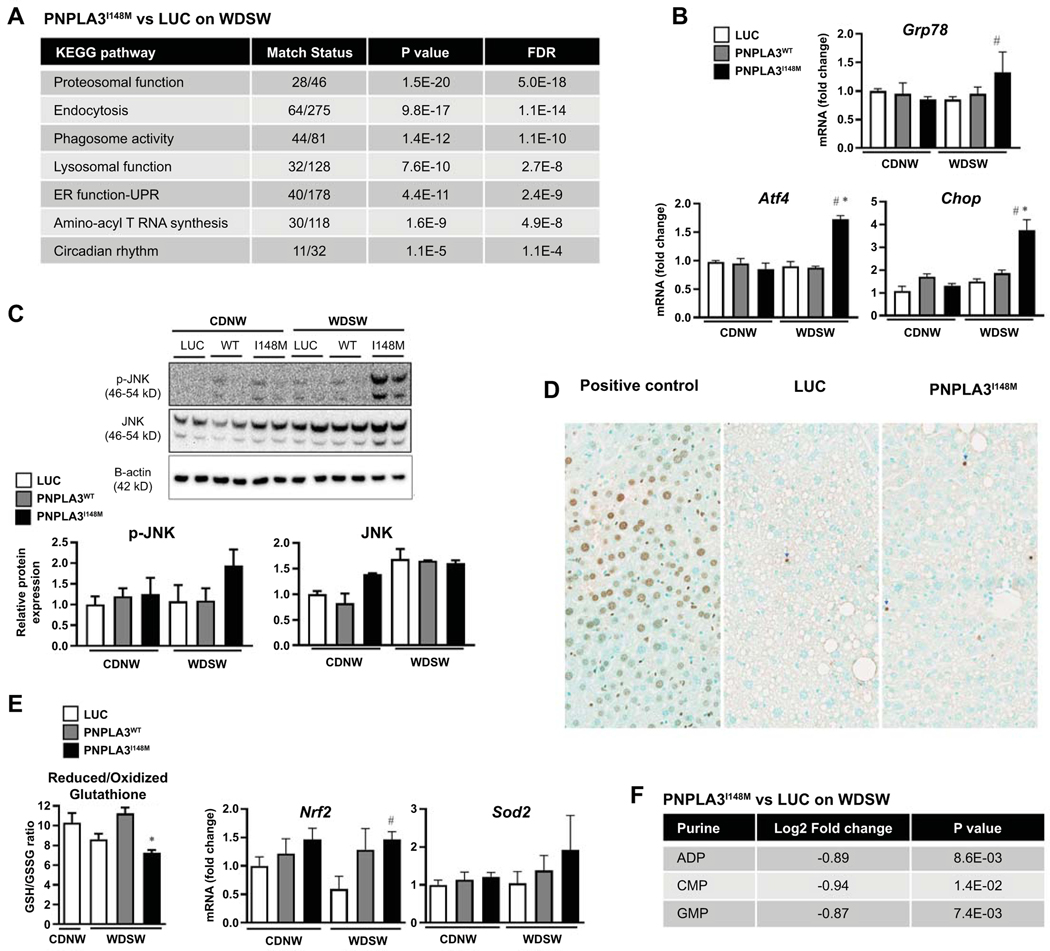
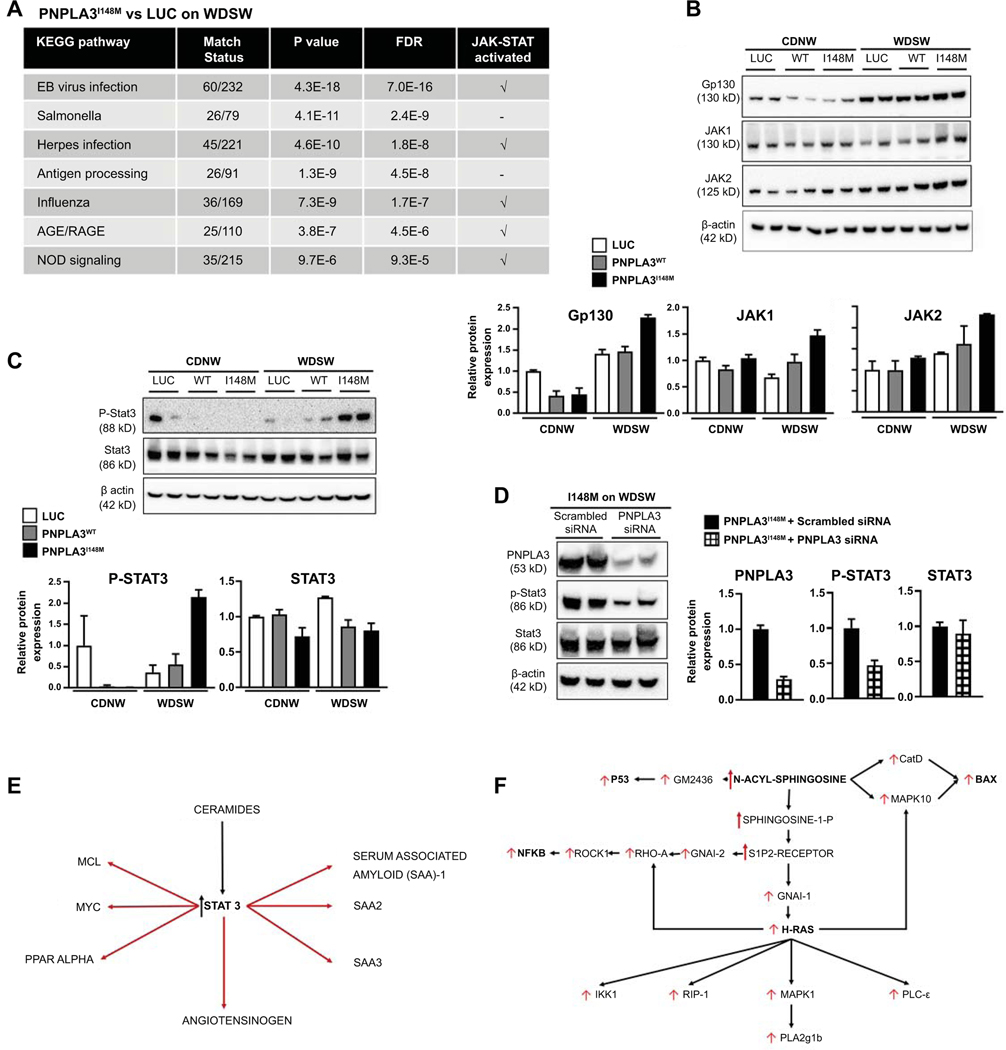
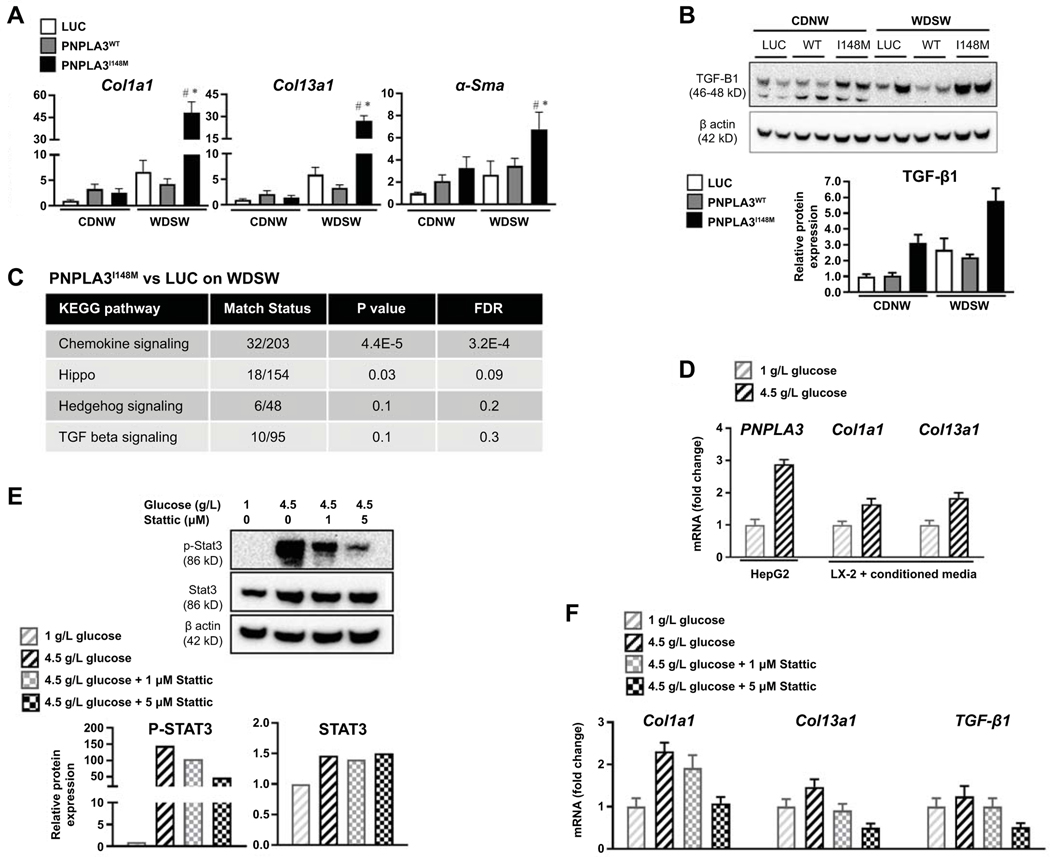
Approach and results: Hepatocyte-specific overexpression of empty vector (luciferase), human wild-type PNPLA3, or PNPLA3I148M was achieved using adeno-associated virus 8 in a diet-induced mouse model of nonalcoholic fatty liver disease followed by chow diet or high-fat Western diet with ad libitum administration of sugar in drinking water (WDSW) for 8 weeks. Under WDSW, PNPLA3I148M overexpression accelerated steatohepatitis with increased steatosis, inflammation ballooning, and fibrosis (P < 0.001 versus other groups for all). Silencing PNPLA3I148M after its initial overexpression abrogated these findings. PNPLA3I148M caused 22:6n3 docosahexanoic acid depletion and increased ceramides under WDSW in addition to increasing triglycerides and diglycerides, especially enriched with unsaturated fatty acids. It also increased oxidative stress and endoplasmic reticulum stress. Increased total ceramides was associated with signature of transducer and activator of transcription 3 (STAT3) activation with downstream activation of multiple immune-inflammatory pathways at a transcriptomic level by network analyses. Silencing PNPLA3I148M reversed STAT3 activation. Conditioned media from HepG2 cells overexpressing PNPLA3I148M increased procollagen mRNA expression in LX2 cells; this was abrogated by hepatocyte STAT3 inhibition.
Conclusions: Under WDSW, PNPLA3I148M overexpression promotes steatosis and NASH by metabolic reprogramming characterized by increased triglycerides and diglycerides, n3 polyunsaturated fatty acid depletion, and increased ceramides with resultant STAT3 phosphorylation and downstream inflammatory pathway activation driving increased stellate cell fibrogenic activity.
© 2020 by the American Association for the Study of Liver Diseases.
Figures
Similar articles
-
Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice.Mol Metab. 2019 Apr;22:49-61. doi: 10.1016/j.molmet.2019.01.013. Epub 2019 Feb 5. Mol Metab. 2019. PMID: 30772256 Free PMC article.
-
PNPLA3 I148M variant in nonalcoholic fatty liver disease: demographic and ethnic characteristics and the role of the variant in nonalcoholic fatty liver fibrosis.World J Gastroenterol. 2015 Jan 21;21(3):794-802. doi: 10.3748/wjg.v21.i3.794. World J Gastroenterol. 2015. PMID: 25624712 Free PMC article. Review.
-
A novel human hepatocyte cell line to study PNPLA3-associated steatotic liver disease.Am J Physiol Gastrointest Liver Physiol. 2025 Jul 1;329(1):G1-G16. doi: 10.1152/ajpgi.00193.2024. Epub 2025 Apr 23. Am J Physiol Gastrointest Liver Physiol. 2025. PMID: 40266007
-
Long-term hypercaloric diet exacerbates metabolic liver disease in PNPLA3 I148M animals.Liver Int. 2023 Aug;43(8):1699-1713. doi: 10.1111/liv.15587. Epub 2023 Apr 18. Liver Int. 2023. PMID: 37073116
-
PNPLA3 I148M polymorphism and progressive liver disease.World J Gastroenterol. 2013 Nov 7;19(41):6969-78. doi: 10.3748/wjg.v19.i41.6969. World J Gastroenterol. 2013. PMID: 24222941 Free PMC article. Review.
Cited by
-
Hepatic patatin-like phospholipase domain-containing 3 levels are increased in I148M risk allele carriers and correlate with NAFLD in humans.Hepatol Commun. 2022 Oct;6(10):2689-2701. doi: 10.1002/hep4.2032. Epub 2022 Jul 14. Hepatol Commun. 2022. PMID: 35833455 Free PMC article.
-
Advances in genetic variation in metabolism-related fatty liver disease.Front Genet. 2023 Sep 11;14:1213916. doi: 10.3389/fgene.2023.1213916. eCollection 2023. Front Genet. 2023. PMID: 37753315 Free PMC article. Review.
-
New uses for an old remedy: Digoxin as a potential treatment for steatohepatitis and other disorders.World J Gastroenterol. 2023 Mar 28;29(12):1824-1837. doi: 10.3748/wjg.v29.i12.1824. World J Gastroenterol. 2023. PMID: 37032732 Free PMC article. Review.
-
Precision medicine and nucleotide-based therapeutics to treat steatotic liver disease.Clin Mol Hepatol. 2025 Feb;31(Suppl):S76-S93. doi: 10.3350/cmh.2024.0438. Epub 2024 Aug 5. Clin Mol Hepatol. 2025. PMID: 39103998 Free PMC article. Review.
-
Gene-based therapies for steatotic liver disease.Mol Ther. 2025 Jun 4;33(6):2527-2547. doi: 10.1016/j.ymthe.2025.04.024. Epub 2025 Apr 19. Mol Ther. 2025. PMID: 40254880 Review.
References
-
- Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010;51:1209–1217. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
