

Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome
- PMID: 33131181
- PMCID: PMC7756669
- DOI: 10.1002/humu.24127
Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome
Abstract
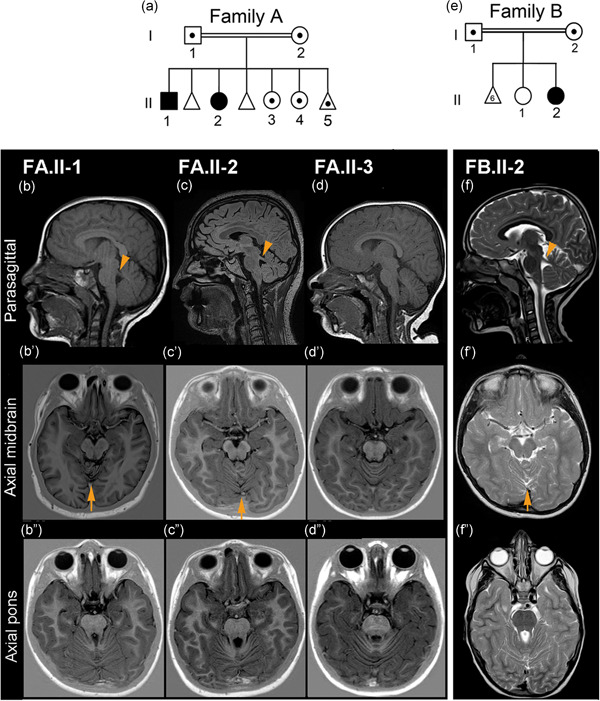
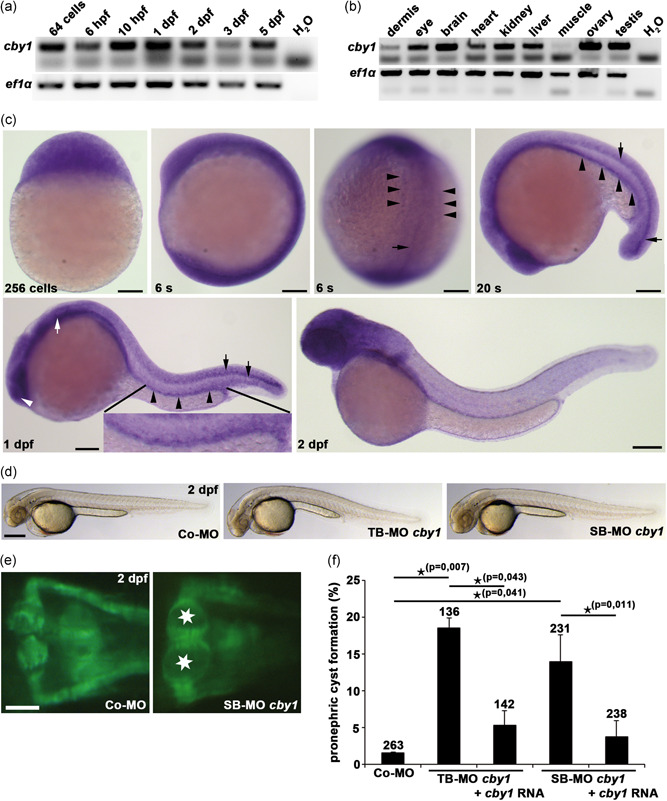
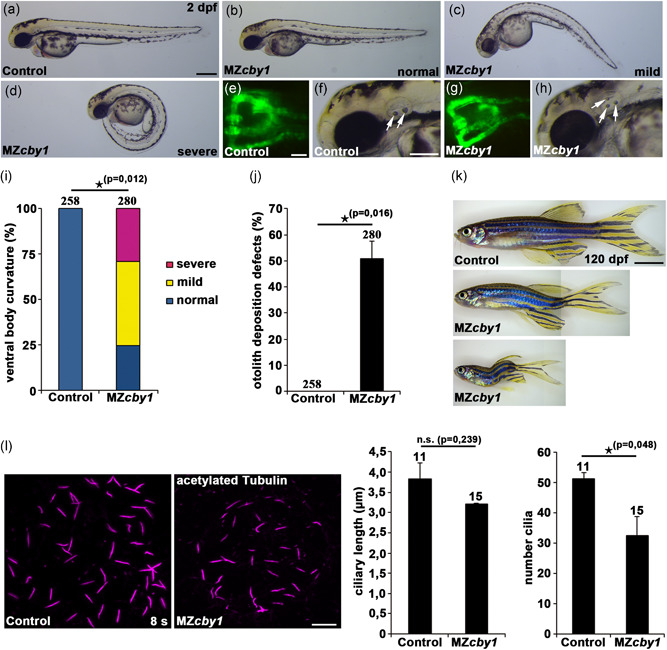
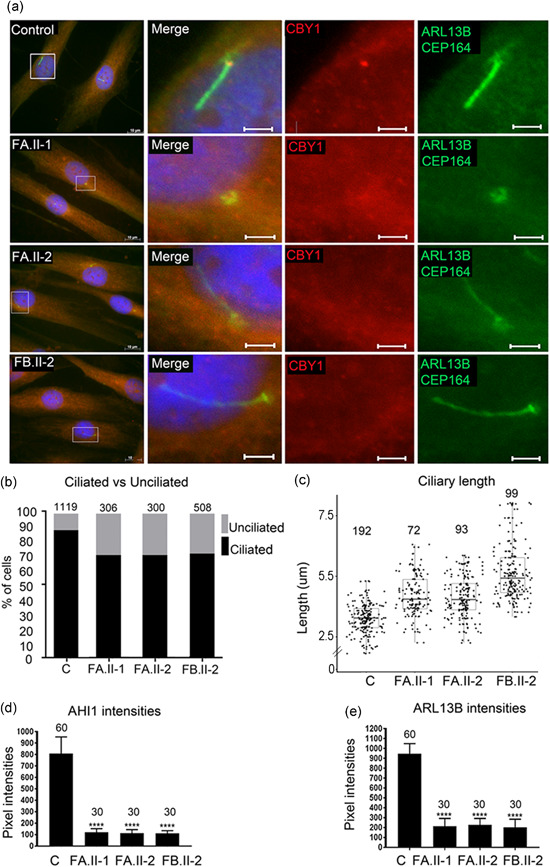
Ciliopathies are clinically and genetically heterogeneous diseases. We studied three patients from two independent families presenting with features of Joubert syndrome: abnormal breathing pattern during infancy, developmental delay/intellectual disability, cerebellar ataxia, molar tooth sign on magnetic resonance imaging scans, and polydactyly. We identified biallelic loss-of-function (LOF) variants in CBY1, segregating with the clinical features of Joubert syndrome in the families. CBY1 localizes to the distal end of the mother centriole, contributing to the formation and function of cilia. In accordance with the clinical and mutational findings in the affected individuals, we demonstrated that depletion of Cby1 in zebrafish causes ciliopathy-related phenotypes. Levels of CBY1 transcript were found reduced in the patients compared with controls, suggesting degradation of the mutated transcript through nonsense-mediated messenger RNA decay. Accordingly, we could detect CBY1 protein in fibroblasts from controls, but not from patients by immunofluorescence. Furthermore, we observed reduced ability to ciliate, increased ciliary length, and reduced levels of the ciliary proteins AHI1 and ARL13B in patient fibroblasts. Our data show that CBY1 LOF-variants cause a ciliopathy with features of Joubert syndrome.
Keywords: CBY1; Joubert syndrome; ciliopathy; primary cilia defect; whole exome sequencing; zebrafish.
© 2020 The Authors. Human Mutation published by Wiley Periodicals LLC.
Conflict of interest statement
Nicholas Katsanis is a paid consultant for and holds significant stock of Rescindo Therapeutics, Inc. Eva Decker, Nadine Bachmann, and Carsten Bergmann are employees of the Limbach group for which Carsten Bergmann heads and manages Limbach Genetics. In addition, Carsten Bergmann holds a part‐time faculty appointment at the University of Freiburg. The remaining authors declare that there are no conflict of interests.
Figures
References
-
- Aguilar, A. , Meunier, A. , Strehl, L. , Martinovic, J. , Bonniere, M. , Attie‐Bitach, T. , Encha‐Razavi, F. , & Spassky, N. (2012). Analysis of human samples reveals impaired SHH‐dependent cerebellar development in Joubert syndrome/Meckel syndrome. Proceedings of the National Academy of Sciences of the United States of America, 109(42), 16951–16956. 10.1073/pnas.1201408109 - DOI - PMC - PubMed
-
- Bachmann‐Gagescu, R. , Dempsey, J. C. , Phelps, I. G. , O'Roak, B. J. , Knutzen, D. M. , Rue, T. C. , Ishak, G. E. , Isabella, C. R. , Gorden, N. , Adkins, J. , Boyle, E. A. , de Lacy, N. , O'Day, D. , Alswaid, A. , Devi, A. R. , Lingappa, L. , Lourenço, C. , Martorell, L. , Garcia‐Cazorla, À. , … Doherty, D. (2015). Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. Journal of Medical Genetics, 52(8), 514–522. 10.1136/jmedgenet-2015-103087 - DOI - PMC - PubMed
-
- Burke, M. C. , Li, F. Q. , Cyge, B. , Arashiro, T. , Brechbuhl, H. M. , Chen, X. , Siller, S. S. , Weiss, M. A. , O'Connell, C. B. , Love, D. , Westlake, C. J. , Reynolds, S. D. , Kuriyama, R. , & Takemaru, K. I. (2014). Chibby promotes ciliary vesicle formation and basal body docking during airway cell differentiation. Journal of Cell Biology, 207(1), 123–137. 10.1083/jcb.201406140 - DOI - PMC - PubMed
-
- Cingolani, P. , Platts, A. , Wang, L. L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S. J. , Lu, X. , & Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin), 6(2), 80–92. 10.4161/fly.19695 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
