

An in-silico analysis of ivermectin interaction with potential SARS-CoV-2 targets and host nuclear importin α
- PMID: 33131430
- PMCID: PMC7643422
- DOI: 10.1080/07391102.2020.1841028
An in-silico analysis of ivermectin interaction with potential SARS-CoV-2 targets and host nuclear importin α
Abstract
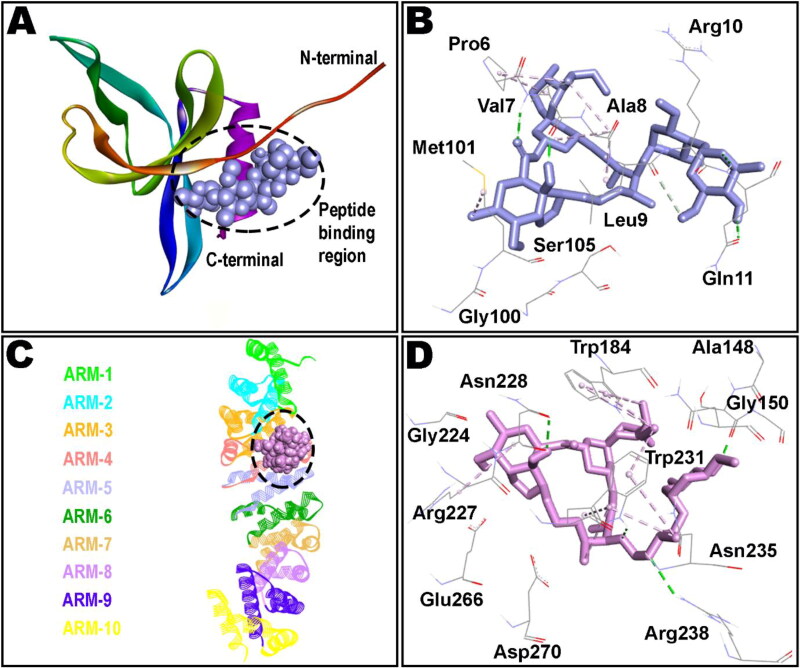
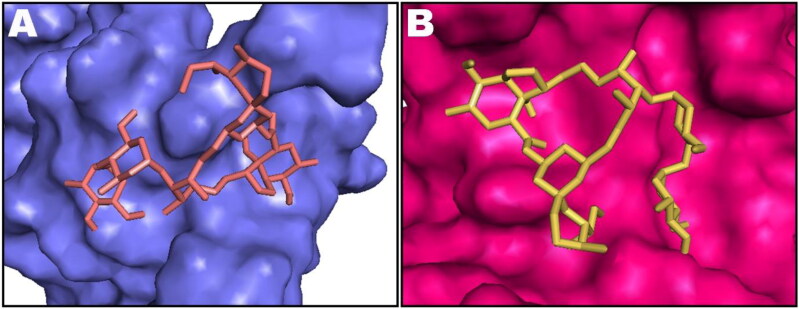
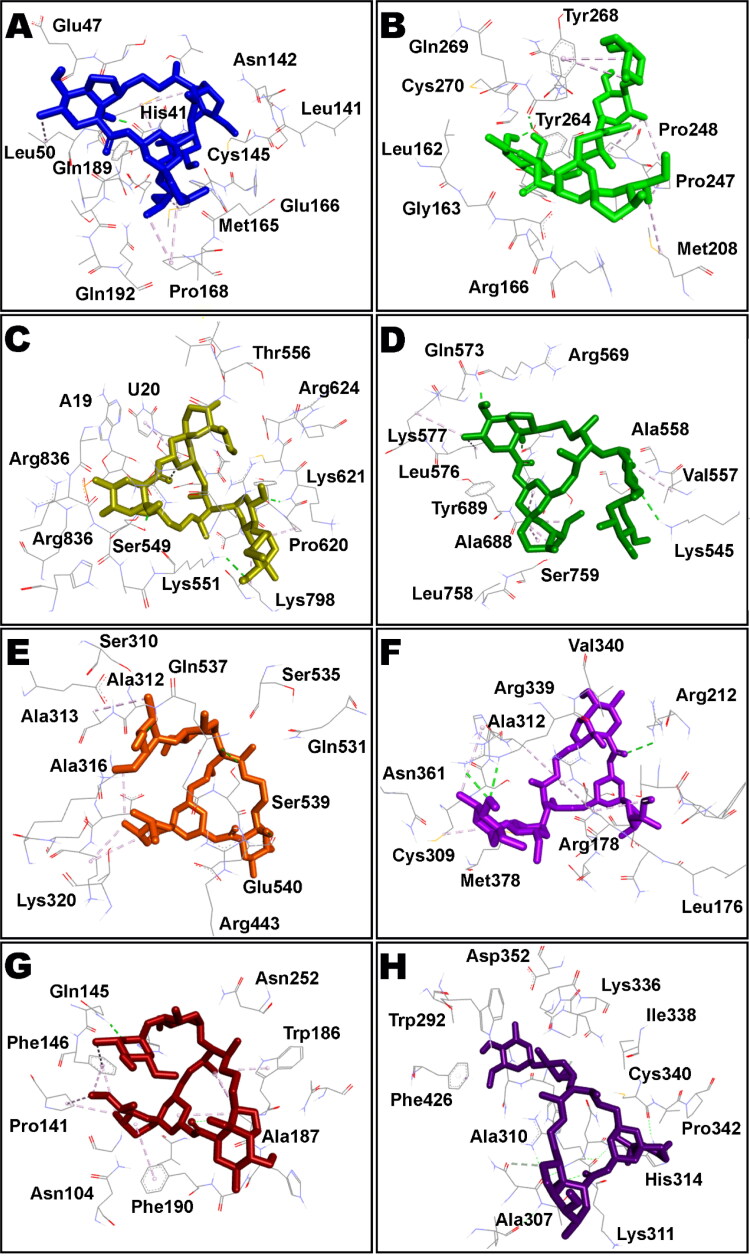
Ivermectin (IVM) is a broad-spectrum antiparasitic agent, having inhibitory potential against wide range of viral infections. It has also been found to hamper SARS-CoV-2 replication in vitro, and its precise mechanism of action against SARS-CoV-2 is yet to be understood. IVM is known to interact with host importin (IMP)α directly and averts interaction with IMPβ1, leading to the prevention of nuclear localization signal (NLS) recognition. Therefore, the current study seeks to employ molecular docking, molecular mechanics generalized Born surface area (MM-GBSA) analysis and molecular dynamics simulation studies for decrypting the binding mode, key interacting residues as well as mechanistic insights on IVM interaction with 15 potential drug targets associated with COVID-19 as well as IMPα. Among all COVID-19 targets, the non-structural protein 9 (Nsp9) exhibited the strongest affinity to IVM showing -5.30 kcal/mol and -84.85 kcal/mol binding energies estimated by AutoDock Vina and MM-GBSA, respectively. However, moderate affinity was accounted for IMPα amounting -6.9 kcal/mol and -66.04 kcal/mol. Stability of the protein-ligand complexes of Nsp9-IVM and IMPα-IVM was ascertained by 100 ns trajectory of all-atom molecular dynamics simulation. Structural conformation of protein in complex with docked IVM exhibited stable root mean square deviation while root mean square fluctuations were also found to be consistent. In silico exploration of the potential targets and their interaction profile with IVM can assist experimental studies as well as designing of COVID-19 drugs. Communicated by Ramaswamy H. Sarma.
Keywords: Antiviral agents; SARS-CoV-2; docking; ivermectin; molecular dynamics.
Conflict of interest statement
No potential conflict of interest was reported by the author.
Figures
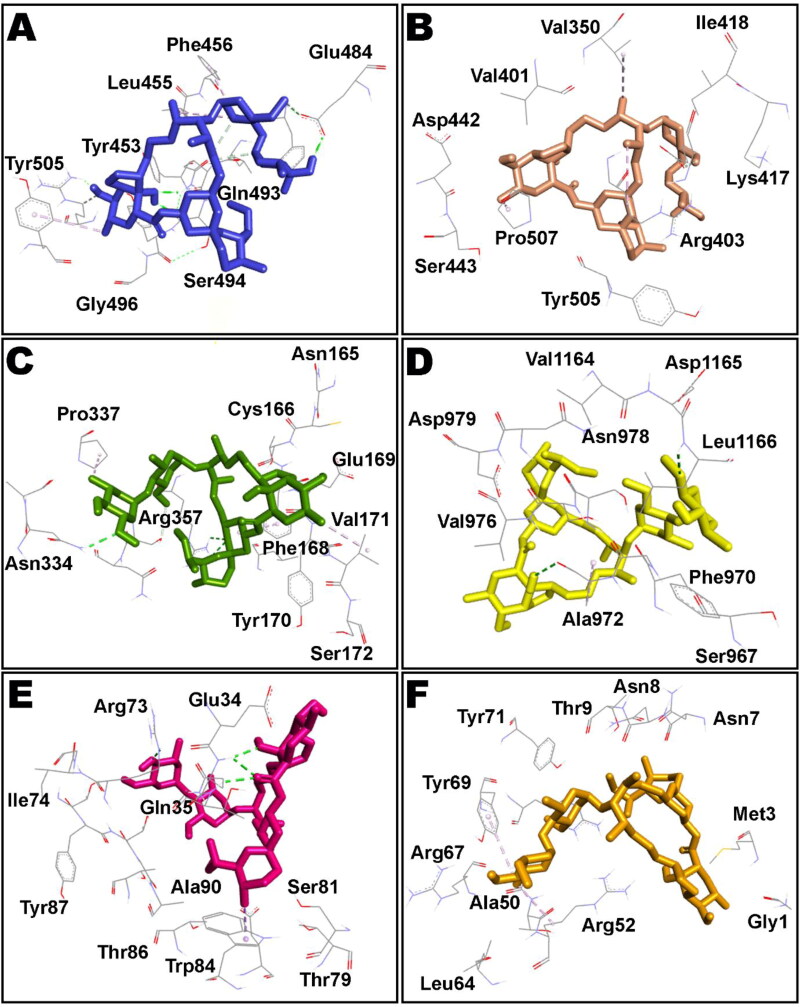
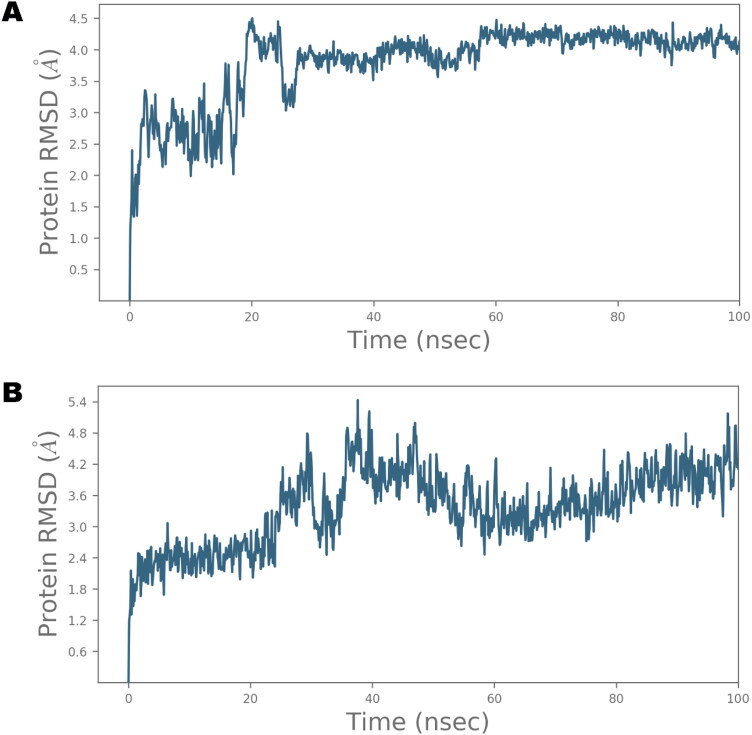
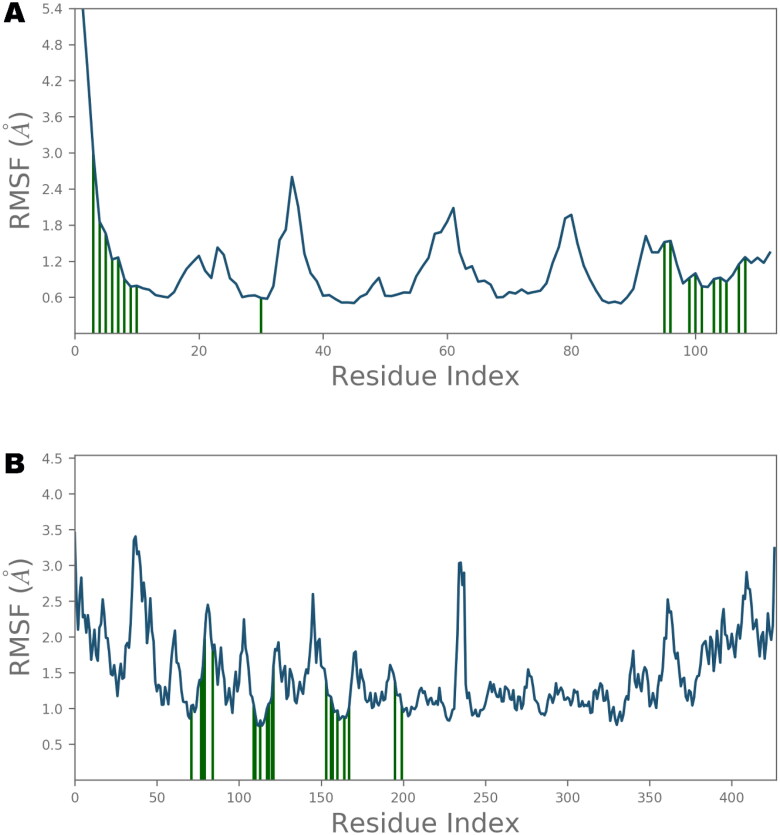
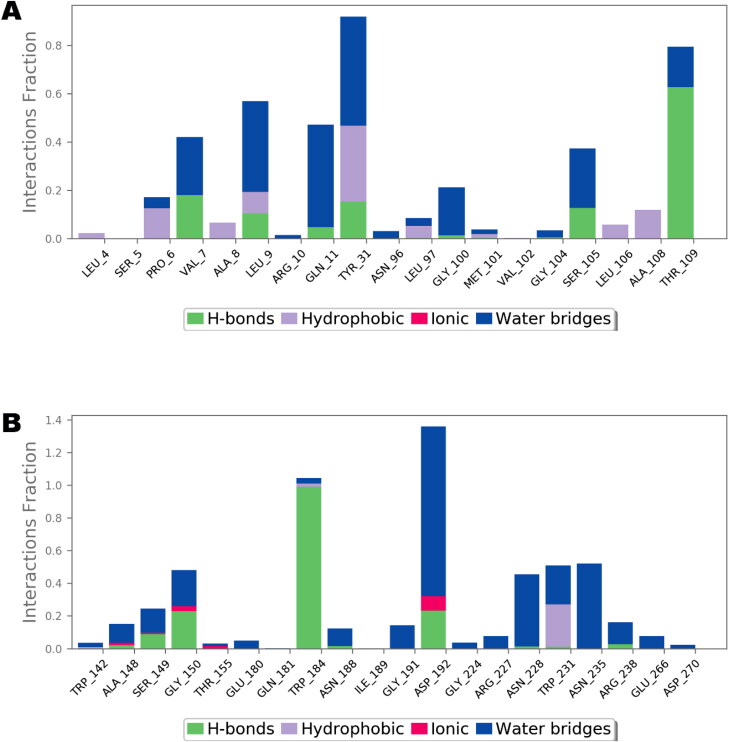
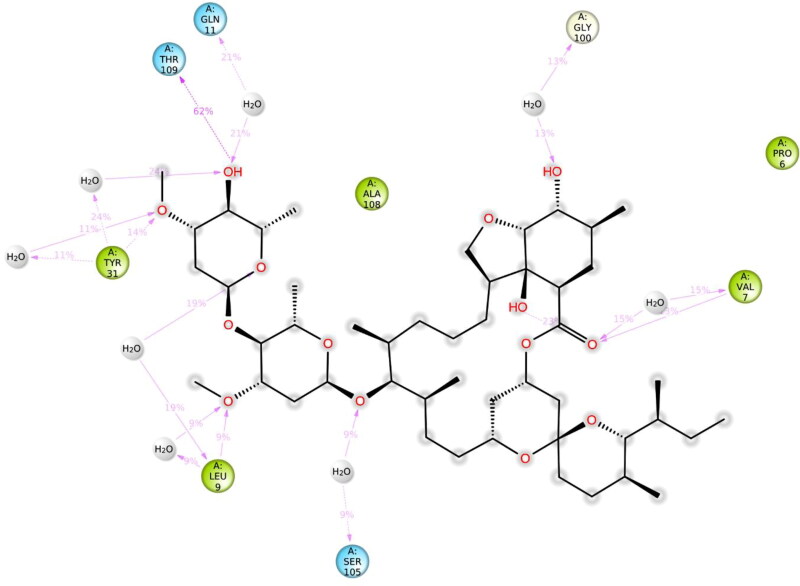
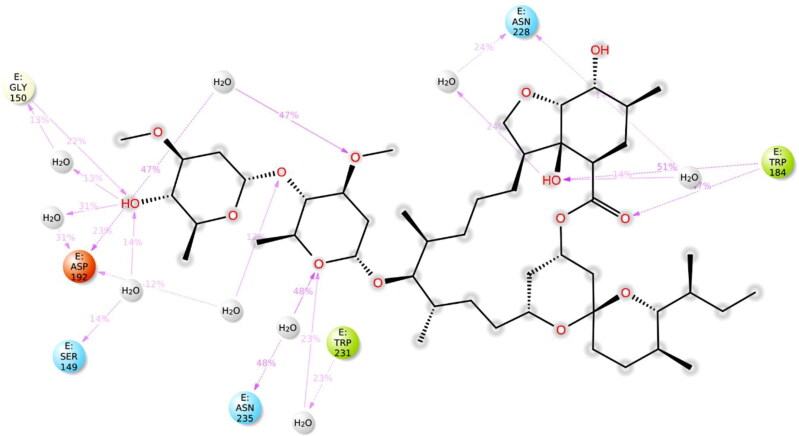
References
-
- Ahmed, M. A., Azam, F., Rghigh, A. M., Gbaj, A., & Zetrini, A. E. (2012). Structure-based design, synthesis, molecular docking, and biological activities of 2-(3-benzoylphenyl) propanoic acid derivatives as dual mechanism drugs. Journal of Pharmacy & Bioallied Sciences, 4(1), 43. 10.4103/0975-7406.92728 - DOI - PMC - PubMed
-
- Ahmed, M., Azam, F., Gbaj, A., Zetrini, A. E., Abodlal, A. S., Rghigh, A., Elmahdi, E., Hamza, A., Salama, M., & Bensaber, S. M. (2016). Ester prodrugs of ketoprofen: Synthesis, in vitro stability, in vivo biological evaluation and in silico comparative docking studies against COX-1 and COX-2. Current Drug Discovery Technologies, 13(1), 41–57. - PubMed
-
- Azam, F., Abodabos, H. S., Taban, I. M., Rfieda, A. R., Mahmood, D., Anwar, M. J., Khan, S., Sizochenko, N., Poli, G., Tuccinardi, T., & Ali, H. I. (2019). Rutin as promising drug for the treatment of Parkinson’s disease: An assessment of MAO-B inhibitory potential by docking, molecular dynamics and DFT studies. Molecular Simulation, 45(18), 1563–1571. 10.1080/08927022.2019.1662003 - DOI
-
- Azam, F., Alabdullah, N. H., Ehmedat, H. M., Abulifa, A. R., Taban, I., & Upadhyayula, S. (2018). NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. Journal of Biomolecular Structure and Dynamics, 36(8), 2099–2117. 10.1080/07391102.2017.1338164 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous