

Screening Malaria-box compounds to identify potential inhibitors against SARS-CoV-2 Mpro, using molecular docking and dynamics simulation studies
- PMID: 33131721
- PMCID: PMC7584923
- DOI: 10.1016/j.ejphar.2020.173664
Screening Malaria-box compounds to identify potential inhibitors against SARS-CoV-2 Mpro, using molecular docking and dynamics simulation studies
Abstract
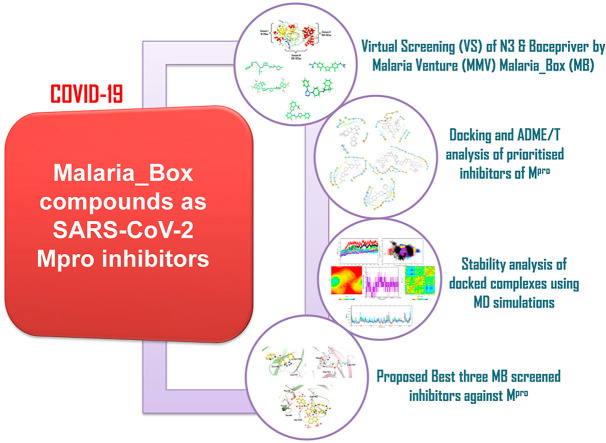
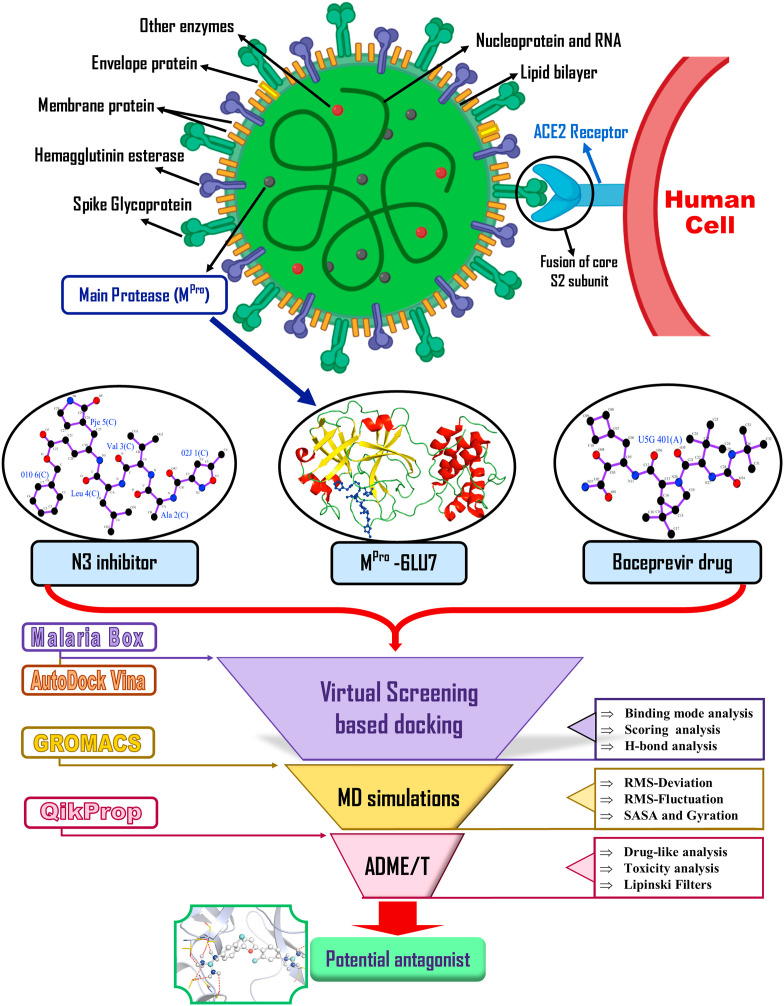
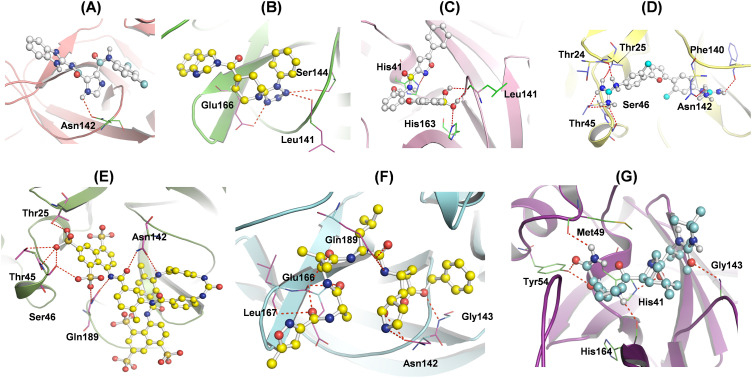
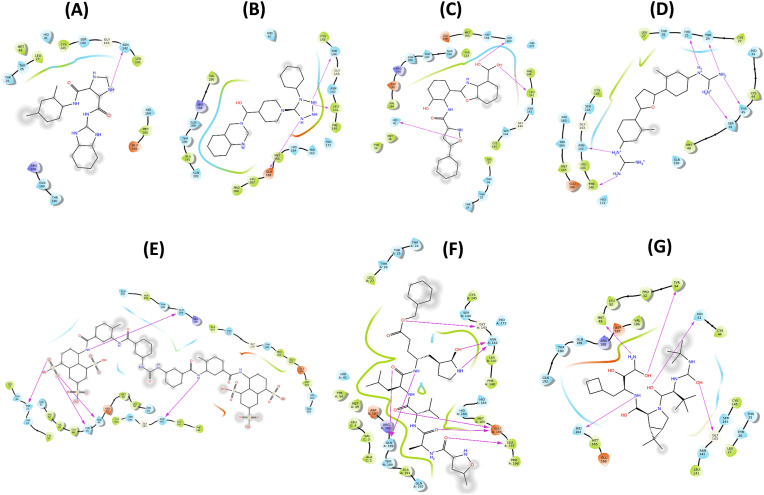
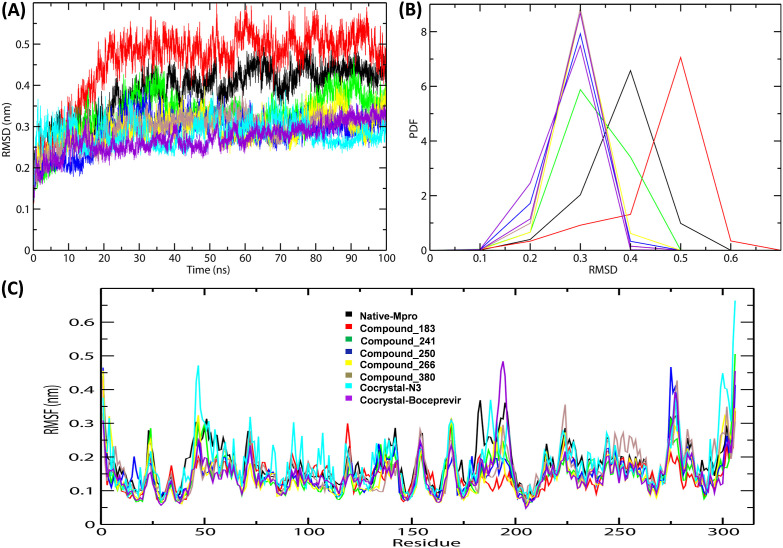
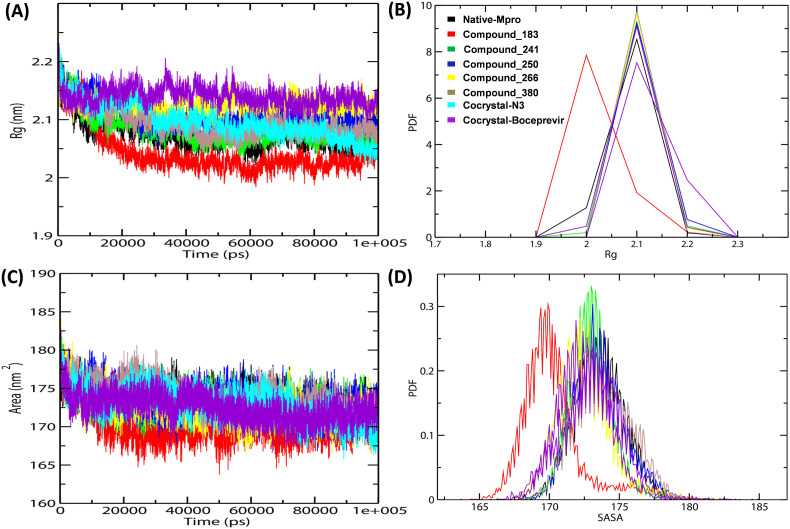
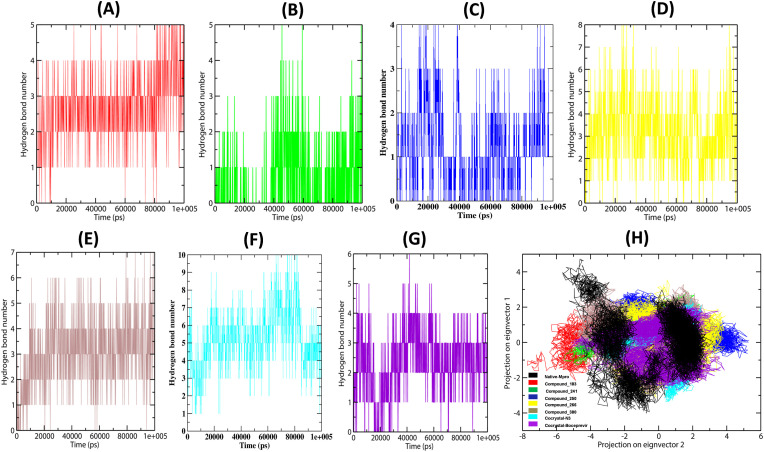
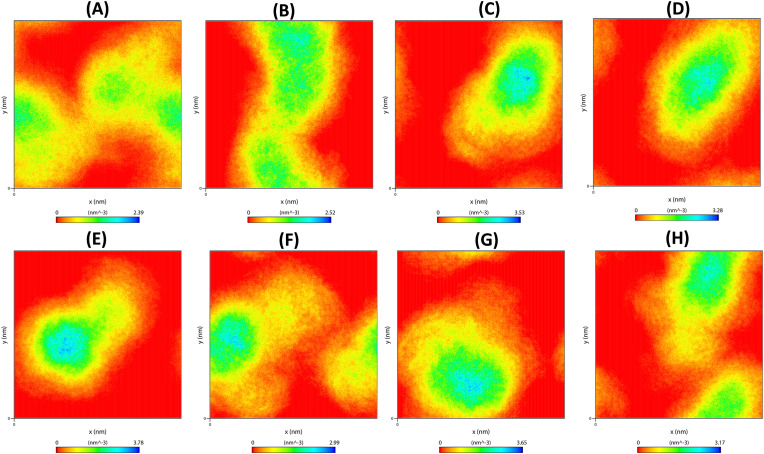
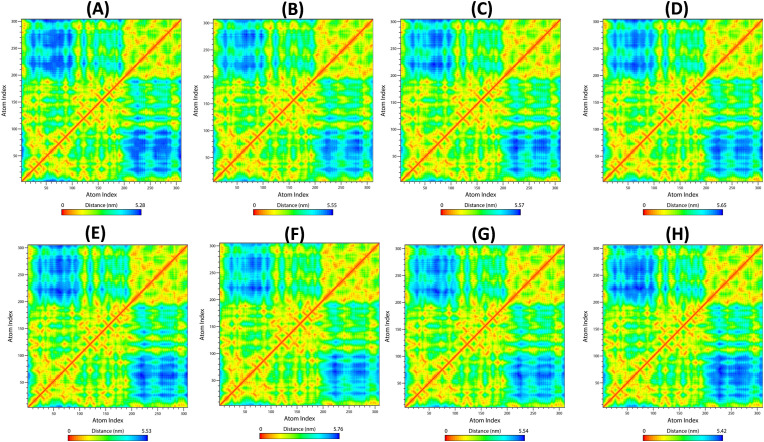
Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) Main protease (Mpro) is one of the vital drug targets amongst all the coronaviruses, as the protein is indispensable for virus replication. The study aimed to identify promising lead molecules against Mpro enzyme through virtual screening of Malaria Venture (MMV) Malaria Box (MB) comprising of 400 experimentally proven compounds. The binding affinities were studied using virtual screening based molecular docking, which revealed five molecules having the highest affinity scores compared to the reference molecules. Utilizing the established 3D structure of Mpro the binding affinity conformations of the docked complexes were studied by Molecular Dynamics (MD) simulations. The MD simulation trajectories were analysed to monitor protein deviation, relative fluctuation, atomic gyration, compactness covariance, residue-residue map and free energy landscapes. Based on the present study outcome, we propose three Malaria_box (MB) compounds, namely, MB_241, MB_250 and MB_266 to be the best lead compounds against Mpro activity. The compounds may be evaluated for their inhibitory activities using experimental techniques.
Keywords: COVID-19; M(pro); MMV Malaria_box; SARS-CoV-2; Virtual screening and MD simulations.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
None of the authors declared conflict of interests.
Figures
References
-
- Abdi H., Williams L.J. Principal component analysis. Wiley Interdiscipl. Rev.: Comput. Stat. 2010;2:433–459.
-
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25.
-
- Ahamad S., Islam A., Ahmad F., Dwivedi N., Hassan M.I. 2/3D-QSAR, molecular docking and MD simulation studies of FtsZ protein targeting benzimidazoles derivatives. Comput. Biol. Chem. 2019;78:398–413. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
