

Detection of Copy Number Variants by Short Multiply Aggregated Sequence Homologies
- PMID: 33132082
- PMCID: PMC7722526
- DOI: 10.1016/j.jmoldx.2020.09.009
Detection of Copy Number Variants by Short Multiply Aggregated Sequence Homologies
Abstract
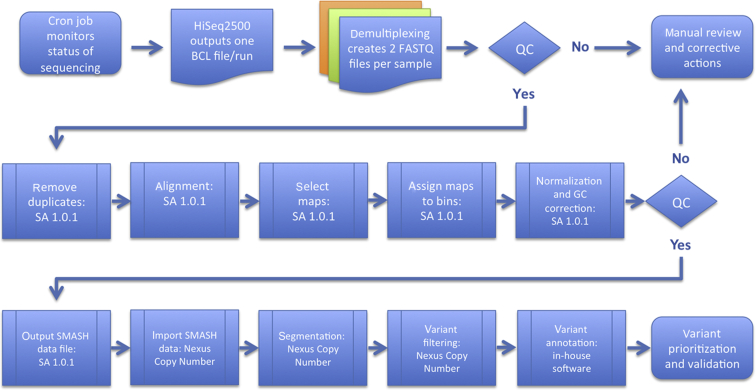
Chromosomal microarray testing is indicated for patients with diagnoses including unexplained developmental delay or intellectual disability, autism spectrum disorders, and multiple congenital anomalies. The short multiply aggregated sequence homologies (SMASH) genomic assay is a novel next-generation sequencing technology that performs copy number analysis at resolution similar to high-coverage whole genome sequencing but requires far less capacity. We benchmarked the performance of SMASH on a panel of genomic DNAs containing known copy number variants (CNVs). SMASH was able to detect pathogenic copy number variants of ≥10 kb in 77 of 77 samples. No pathogenic events were seen in 32 of 32 controls, indicating 100% sensitivity and specificity for detecting pathogenic CNVs >10 kb. Repeatability (interassay precision) and reproducibility (intra-assay precision) were assessed with 13 samples and showed perfect concordance. We also established that SMASH had a limit of detection of 20% for detection of large mosaic CNVs. Finally, we analyzed seven blinded specimens by SMASH analysis and successfully identified all pathogenic events. These results establish the efficacy of the SMASH genomic assay as a clinical test for the detection of pathogenic copy number variants at a resolution comparable to chromosomal microarray analysis.
Copyright © 2020 Association for Molecular Pathology and American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J., Faucett W.A., Feuk L., Friedman J.M., Hamosh A., Jackson L., Kaminsky E.B., Kok K., Krantz I.D., Kuhn R.M., Lee C., Ostell J.M., Rosenberg C., Scherer S.W., Spinner N.B., Stavropoulos D.J., Tepperberg J.H., Thorland E.C., Vermeesch J.R., Waggoner D.J., Watson M.S., Martin C.L., Ledbetter D.H. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. - PMC - PubMed
-
- Trost B., Walker S., Wang Z., Thiruvahindrapuram B., MacDonald J.R., Sung W.W.L., Pereira S.L., Whitney J., Chan A.J.S., Pellecchia G., Reuter M.S., Lok S., Yuen R.K.C., Marshall C.R., Merico D., Scherer S.W. A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet. 2018;102:142–155. - PMC - PubMed
-
- South S.T., Lee C., Lamb A.N., Higgins A.W., Kearney H.M., Working Group for the American College of Medical Genetics. Genomics Laboratory Quality Assurance Committee ACMG standards and guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet Med. 2013;15:901–909. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
