

Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases
- PMID: 33132902
- PMCID: PMC7572858
- DOI: 10.3389/fnagi.2020.581374
Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases
Abstract
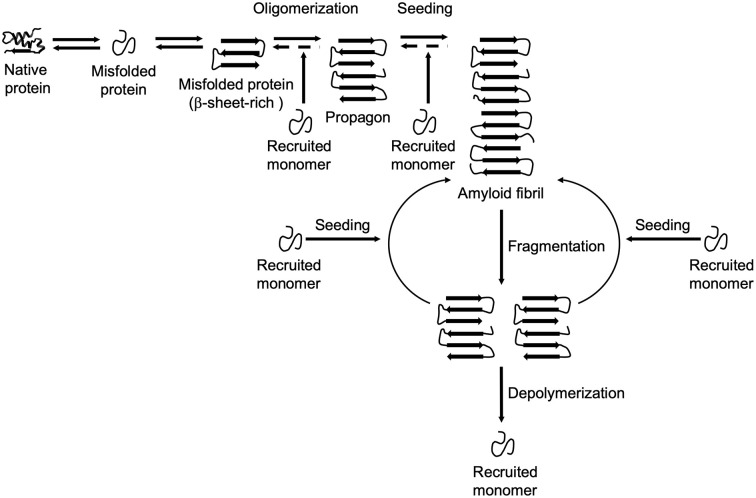
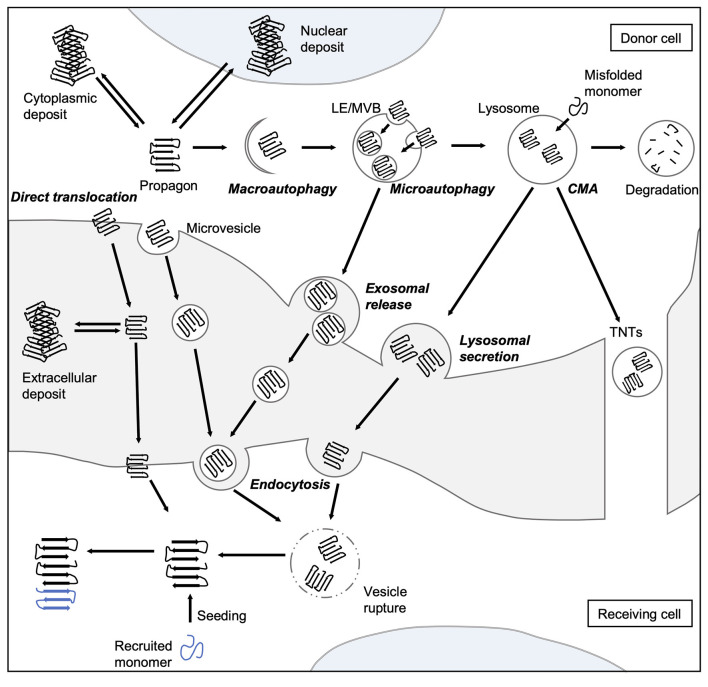
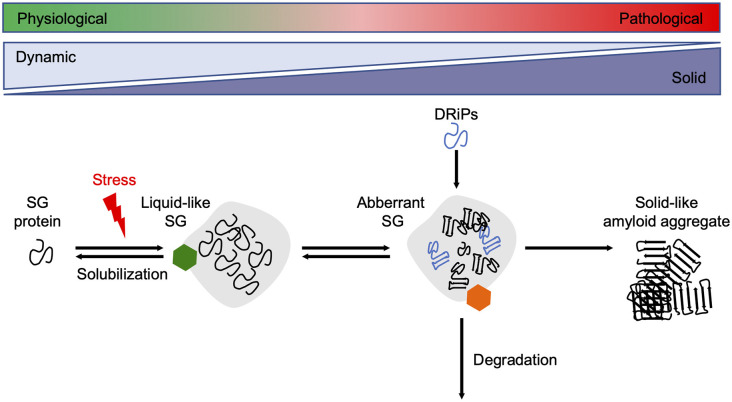
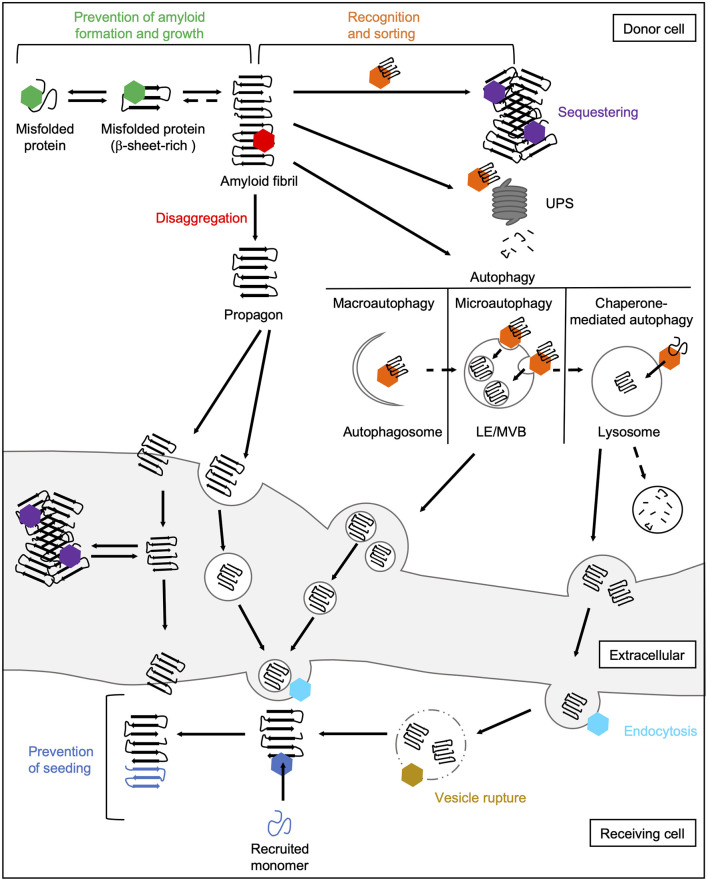
Aberrant accumulation of misfolded proteins into amyloid deposits is a hallmark in many age-related neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS). Pathological inclusions and the associated toxicity appear to spread through the nervous system in a characteristic pattern during the disease. This has been attributed to a prion-like behavior of amyloid-type aggregates, which involves self-replication of the pathological conformation, intercellular transfer, and the subsequent seeding of native forms of the same protein in the neighboring cell. Molecular chaperones play a major role in maintaining cellular proteostasis by assisting the (re)-folding of cellular proteins to ensure their function or by promoting the degradation of terminally misfolded proteins to prevent damage. With increasing age, however, the capacity of this proteostasis network tends to decrease, which enables the manifestation of neurodegenerative diseases. Recently, there has been a plethora of studies investigating how and when chaperones interact with disease-related proteins, which have advanced our understanding of the role of chaperones in protein misfolding diseases. This review article focuses on the steps of prion-like propagation from initial misfolding and self-templated replication to intercellular spreading and discusses the influence that chaperones have on these various steps, highlighting both the positive and adverse consequences chaperone action can have. Understanding how chaperones alleviate and aggravate disease progression is vital for the development of therapeutic strategies to combat these debilitating diseases.
Keywords: disaggregation; molecular chaperones and Hsps; neurodegenarative diseases; prion-like spreading; proteostasis.
Copyright © 2020 Tittelmeier, Nachman and Nussbaum-Krammer.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous