

PPARα Ameliorates Doxorubicin-Induced Cardiotoxicity by Reducing Mitochondria-Dependent Apoptosis via Regulating MEOX1
- PMID: 33132907
- PMCID: PMC7578427
- DOI: 10.3389/fphar.2020.528267
PPARα Ameliorates Doxorubicin-Induced Cardiotoxicity by Reducing Mitochondria-Dependent Apoptosis via Regulating MEOX1
Abstract
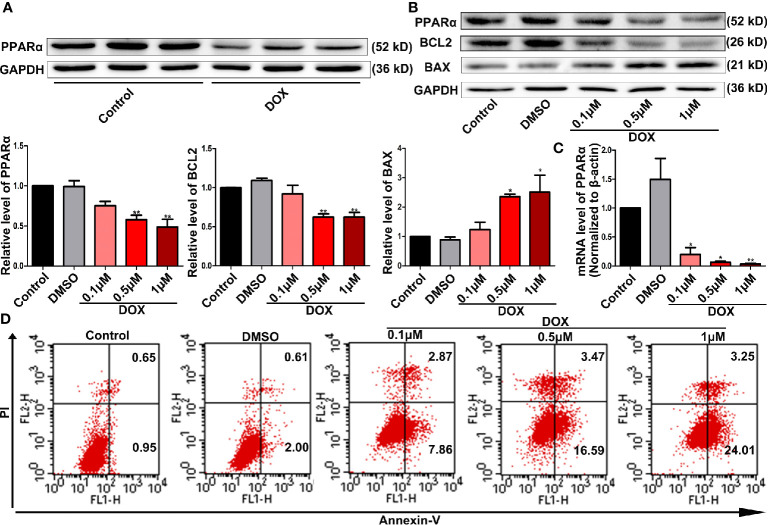
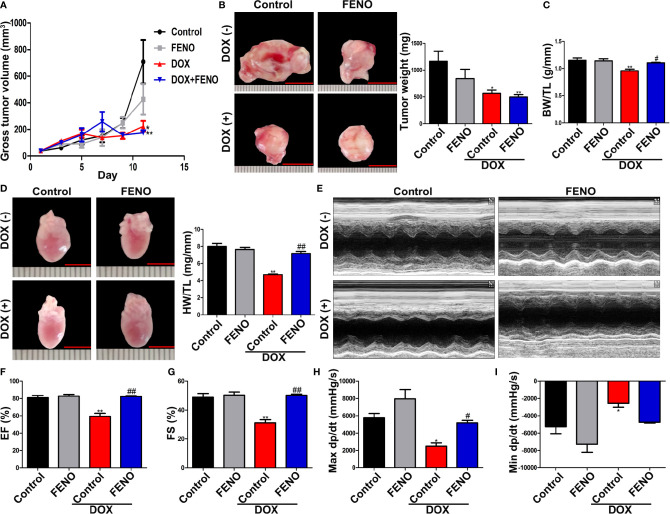
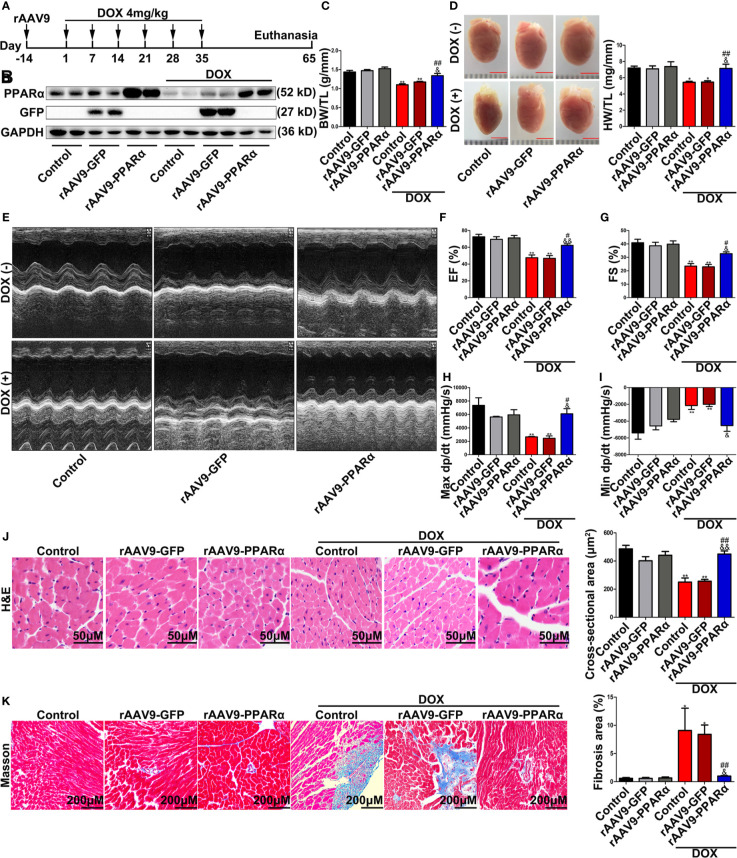
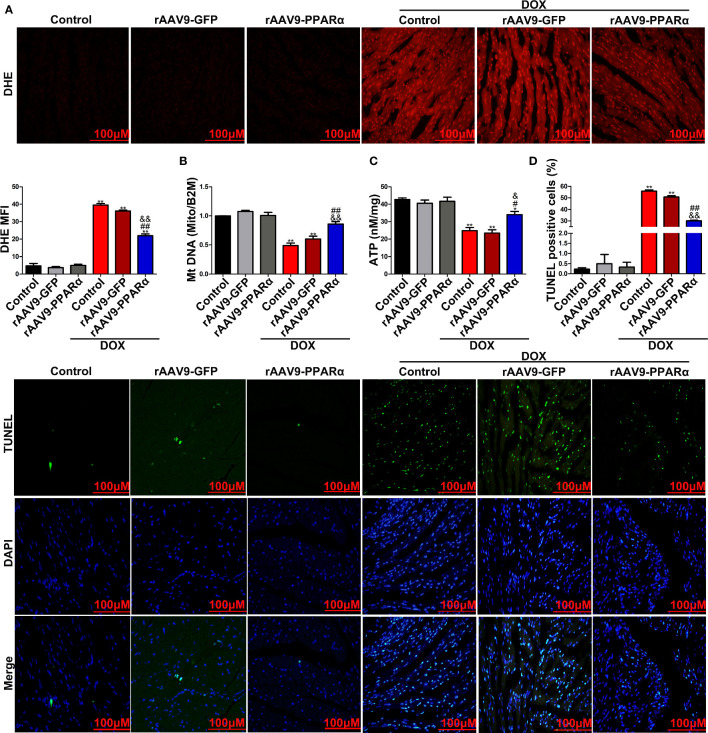
Doxorubicin (DOX), a chemotherapeutic drug widely used in the clinical setting, is known to cause serious cardiotoxicity and greatly reduces the survival rate as well as quality of life of patients receiving chemotherapy. Peroxisome proliferation activated receptor α (PPARα) is a type of ligand activated receptor of the nuclear hormone receptor family that regulates multiple gene expression. Several studies have shown that PPARα has anti-apoptotic and cardio-protective effects. However, its role in DOX-induced cardiotoxicity is rarely reported. In this study, we observed decreased expression of PPARα in the heart of tumor-bearing mice already treated with DOX; however, no such phenomenon was observed in tumor tissues. Next, we observed that the PPARα agonist, fenofibrate (FENO), had no effect on tumor progression; however, it enhanced cardiac function in tumor-bearing mice treated with DOX. Subsequently, recombinant adeno-associated virus serotype 9 (rAAV9) was used to manipulate the expression of PPARα in the heart of DOX-induced mice. Our results showed that PPARα gene delivery reduced cardiac dysfunction and mitochondria-dependent apoptosis in DOX-induced mice. Furthermore, we found that PPARα directly regulated the expression of mesenchyme homeobox 1 (MEOX1). Most importantly, the cardioprotective effects of PPARα could be neutralized by knocking down MEOX1. In summary, PPARα plays a vital role in DOX-induced cardiotoxicity and is a promising treatment target.
Keywords: PPARα; apoptosis; cardiotoxicity; doxorubicin; mitochondria.
Copyright © 2020 Wang, Fang, Zhang, Wang, Wu and Wang.
Figures
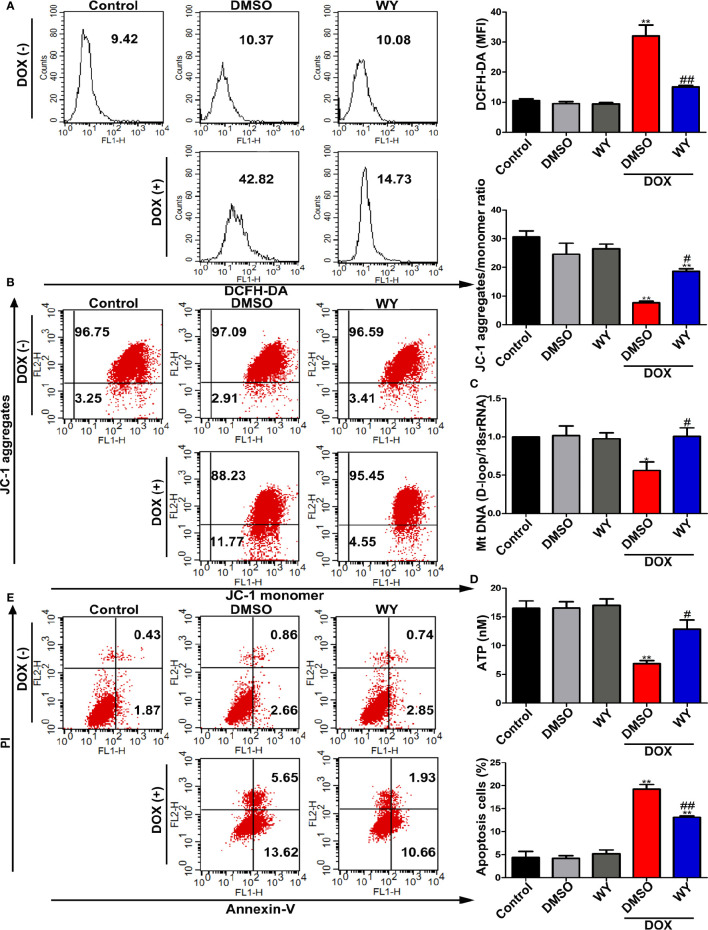
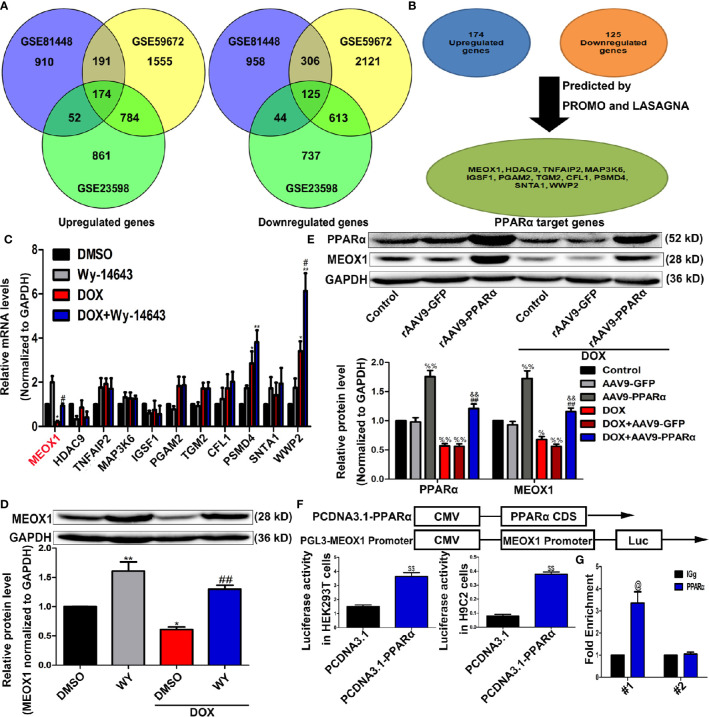
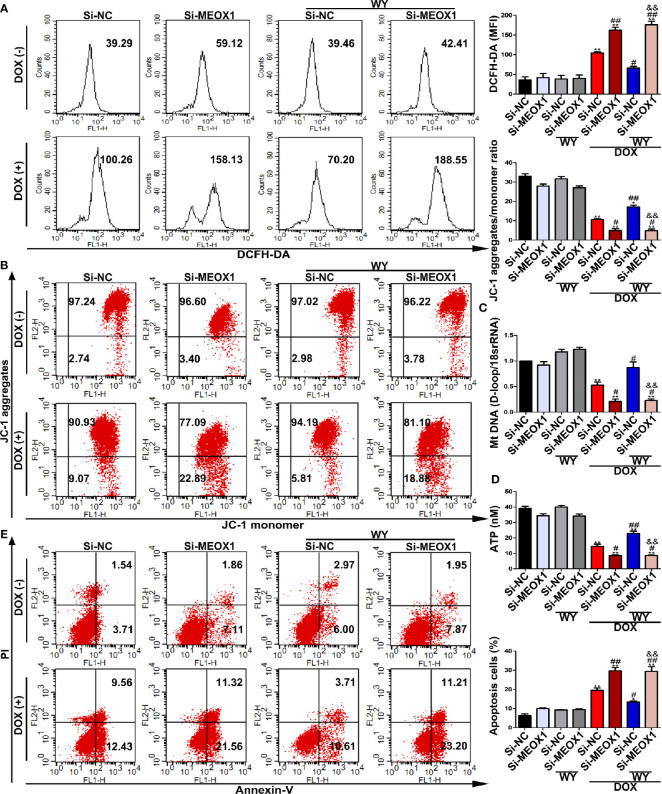
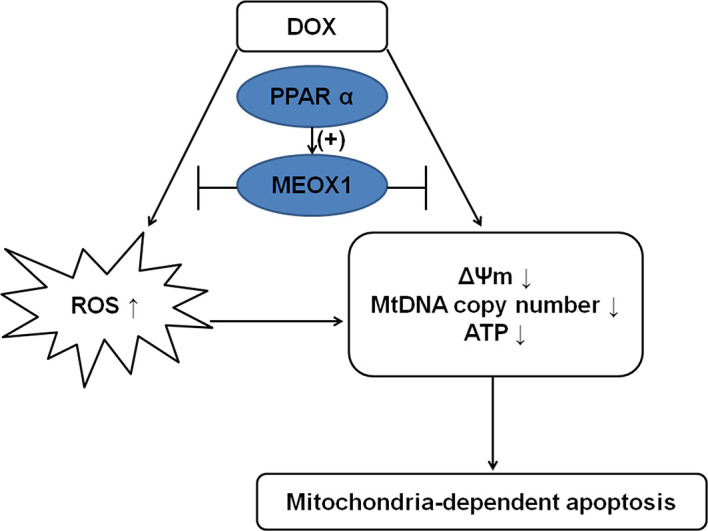
References
LinkOut - more resources
Full Text Sources
Research Materials