

SURF1 related Leigh syndrome: Clinical and molecular findings of 16 patients from Turkey
- PMID: 33134083
- PMCID: PMC7586243
- DOI: 10.1016/j.ymgmr.2020.100657
SURF1 related Leigh syndrome: Clinical and molecular findings of 16 patients from Turkey
Abstract
Introduction: Pathogenic variants in SURF1, a nuclear-encoded gene encoding a mitochondrial chaperone involved in COX assembly, are one of the most common causes of Leigh syndrome (LS).
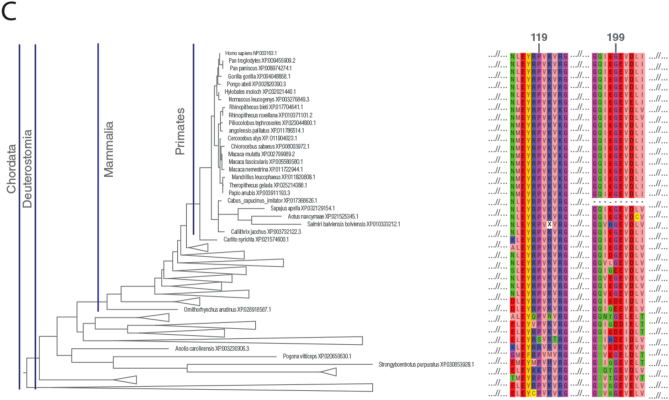
Material-methods: Sixteen patients diagnosed to have SURF1-related LS between 2012 and 2020 were included in the study. Their clinical, biochemical and molecular findings were recorded. 10/16 patients were diagnosed using whole-exome sequencing (WES), 4/16 by Sanger sequencing of SURF1, 1/16 via targeted exome sequencing and 1/16 patient with whole-genome sequencing (WGS). The pathogenicity of SURF1 variants was evaluated by phylogenetic studies and modelling on the 3D structure of the SURF1 protein.
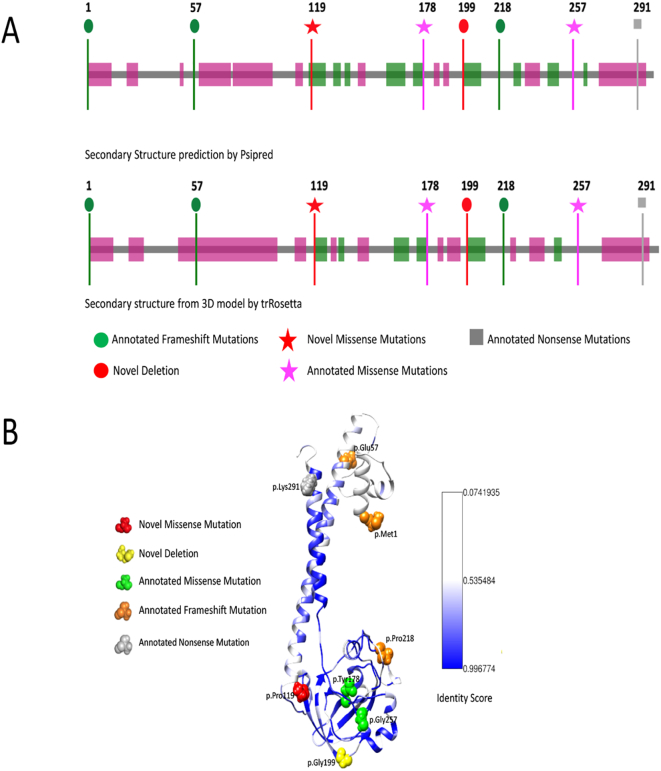
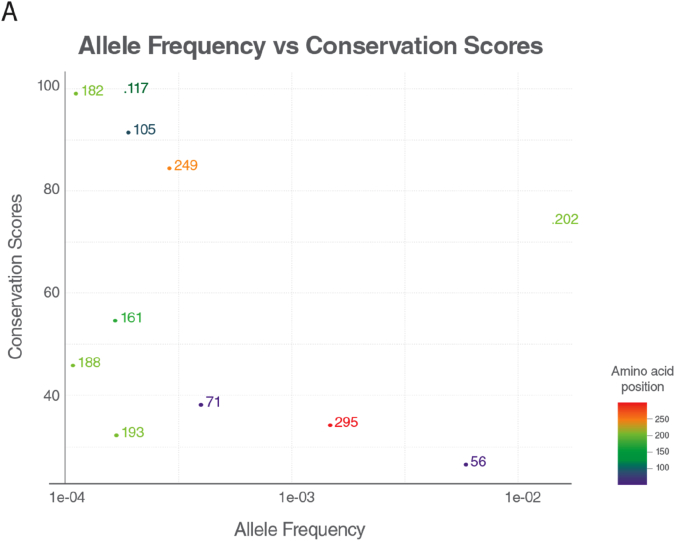
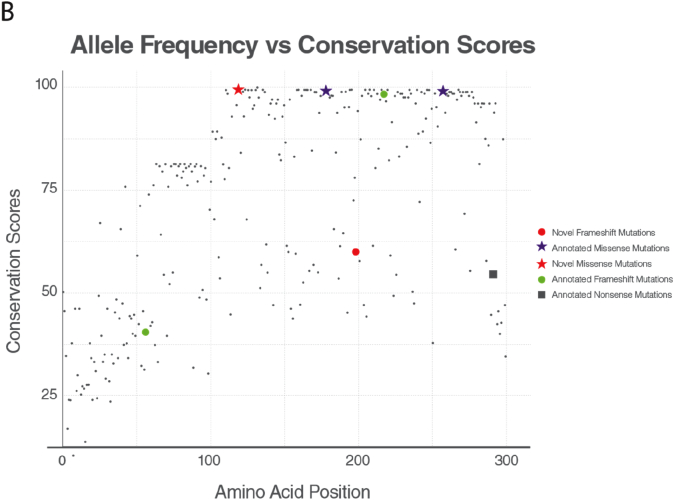
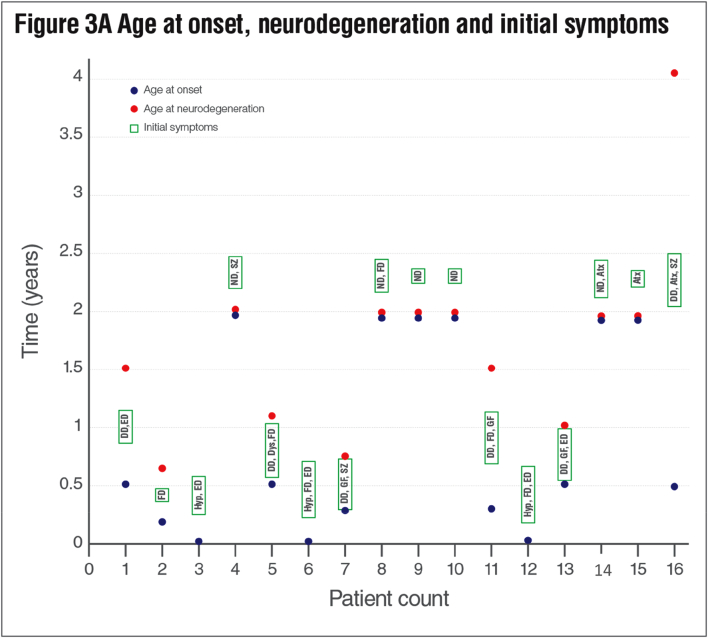
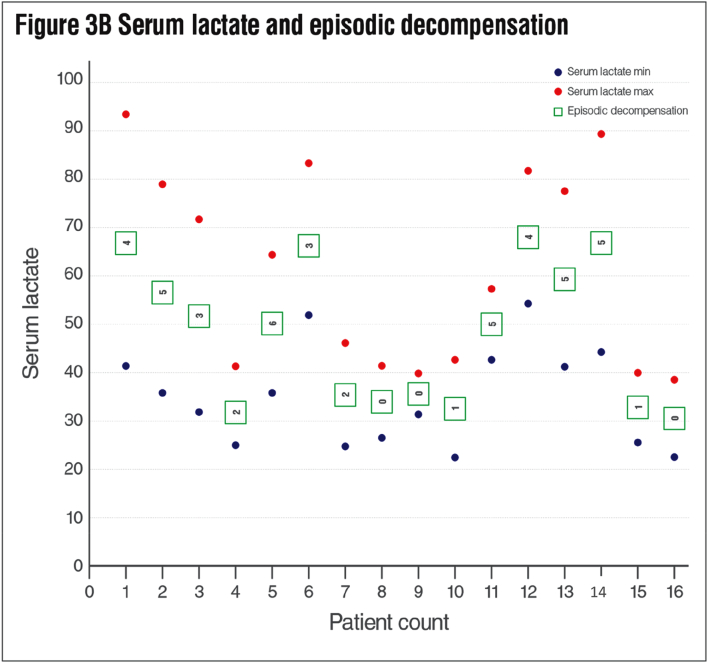
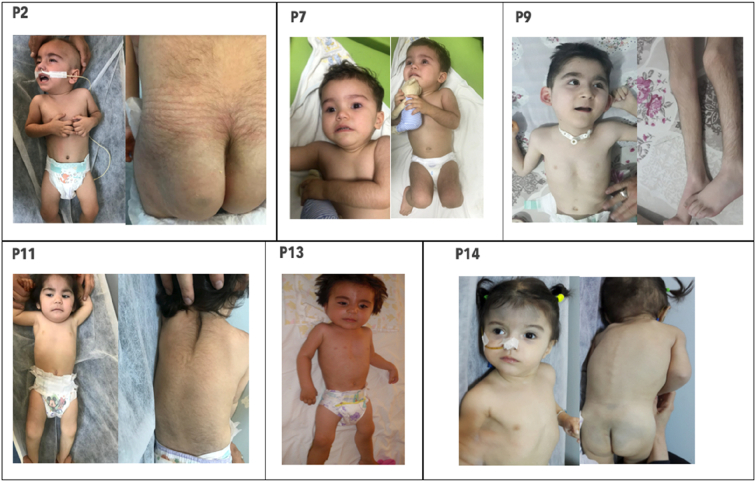
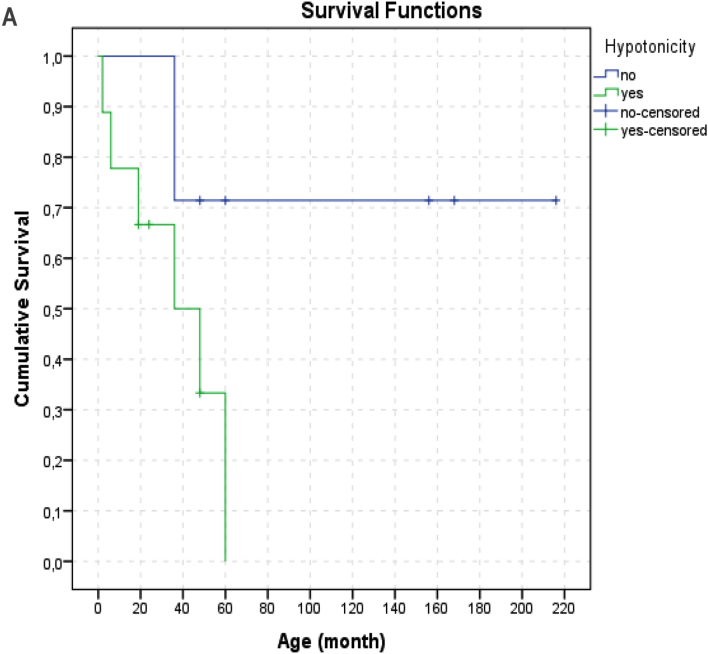
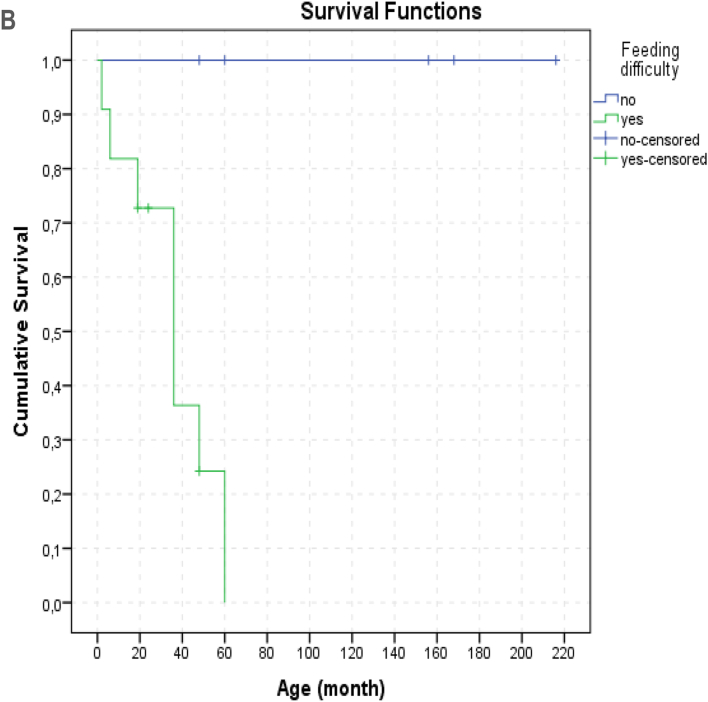
Results: We identified 16 patients from 14 unrelated families who were either homozygous or compound heterozygous for SURF1 pathogenic variants. Nine different SURF1 variants were detected The c.769G > A was the most common variant with an allelic frequency of 42.8% (12/28), c.870dupT [(p.Lys291*); (8/28 28.5%)], c.169delG [(p.Glu57Lysfs*15), (2/24; 7.1%)], c.532 T > A [(p.Tyr178Asn); (2/28, 7.1%)], c.653_654delCT [(p.Pro218Argfs*29); (4/28, 14.2%)] c.595_597delGGA [(p.Gly199del); (1/28, 3.5%)], c.751 + 1G > A (2/28, 4.1%), c.356C > T [(p.Pro119Leu); (2/28, 3.5%)] were the other detected variants. Two pathogenic variants, C.595_597delGGA and c.356C > T, were detected for the first time. The c.769 G > A variant detected in 6 patients from 5 families was evaluated in terms of phenotype-genotype correlation. There was no definite genotype - phenotype correlation.
Conclusions: To date, more than 120 patients of LS with SURF1 pathogenic variants have been reported. We shared the clinical, molecular data and natural course of 16 new SURF1 defect patients from our country. This study is the first comprehensive research from Turkey that provides information about disease-causing variants in the SURF1 gene. The identification of common variants and phenotype of the SURF1 gene is important for understanding SURF1 related LS.
Synopsis: SURF1 gene defects are one of the most important causes of LS; patients have a homogeneous clinical and biochemical phenotype.
Keywords: COX deficiency; Leigh syndrome; Neuroregression; Next-generation sequencing; Nuclear mitochondrial disorders; SURF1 gene.
© 2020 The Authors.
Figures
References
-
- Diniz G., Tosun Yildirim H., Unalp A., Barutcuoglu M., Guzel O., Polat M., Ture S., Ozgonul F., Serdaroglu G. The evaluation of muscle biopsy findings in children with neuromuscular disorders. J. Dr. Behcet Uz Child. Hosp. 2013;2(2):62–67. doi: 10.5222/buchd.2012.062. - DOI
-
- Dınız G., Hazan F., Yildirim H.T., Unalp A., Polat M., Serdaroğlu G., Güzel O., Bağ O., Seçıl Y., Ozgönül F., Türe S., Akhan G., Tükün A. Histopathological and genetic features of patients with limb girdle muscular dystrophy type 2C. Turk Patoloji Dergisi. 2014;30(2):111–117. doi: 10.5146/tjpath.2014.01239. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
