

Rapid in silico Design of Potential Cyclic Peptide Binders Targeting Protein-Protein Interfaces
- PMID: 33134275
- PMCID: PMC7578414
- DOI: 10.3389/fchem.2020.573259
Rapid in silico Design of Potential Cyclic Peptide Binders Targeting Protein-Protein Interfaces
Abstract
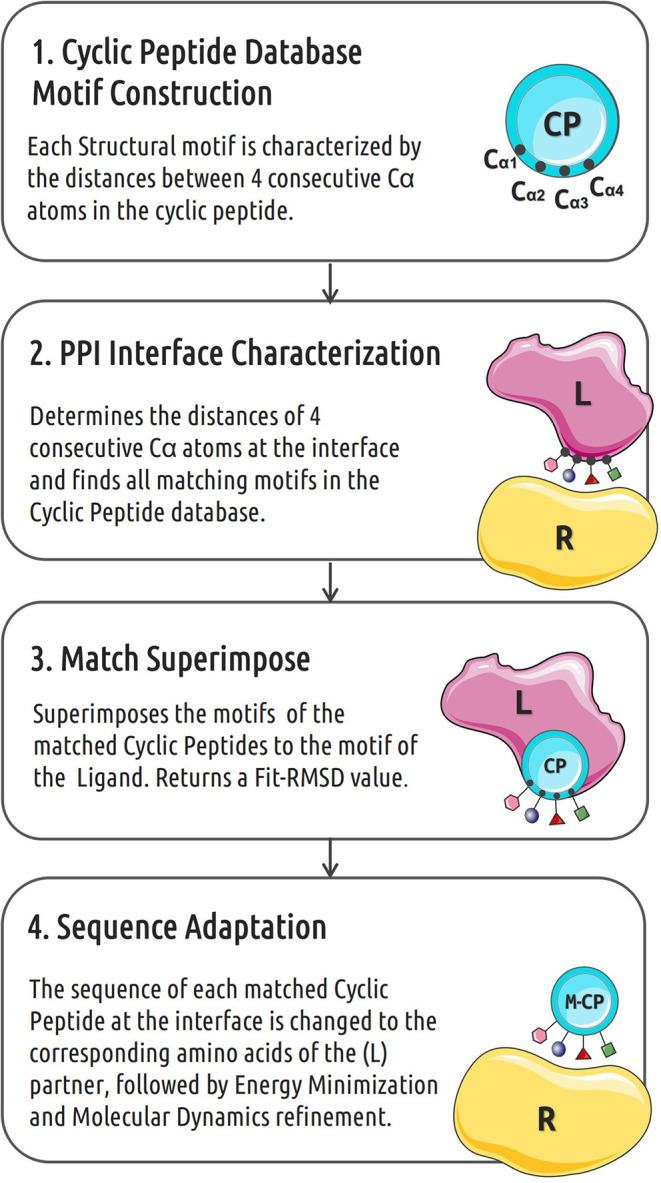
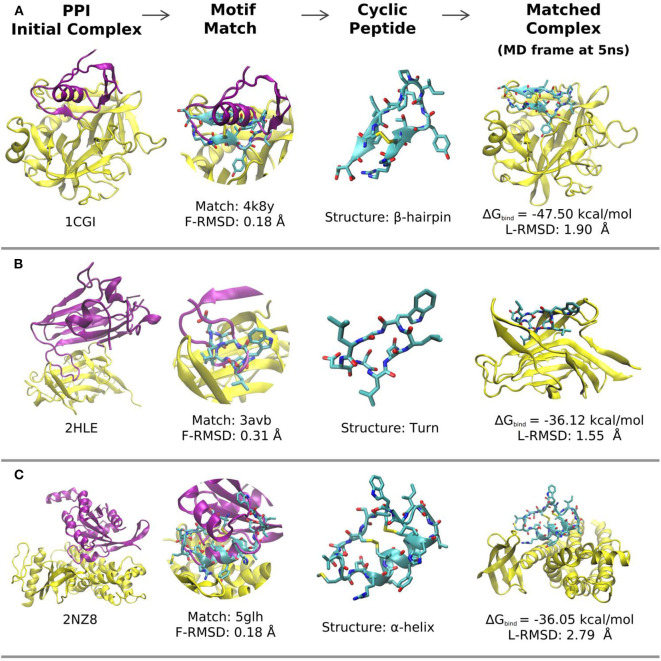
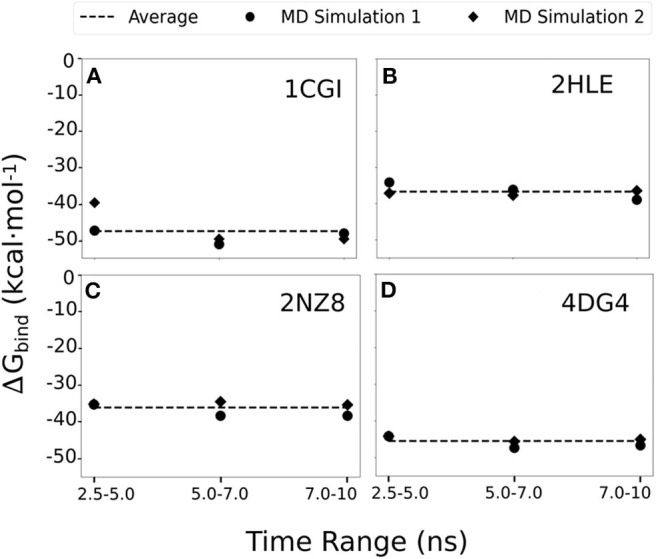
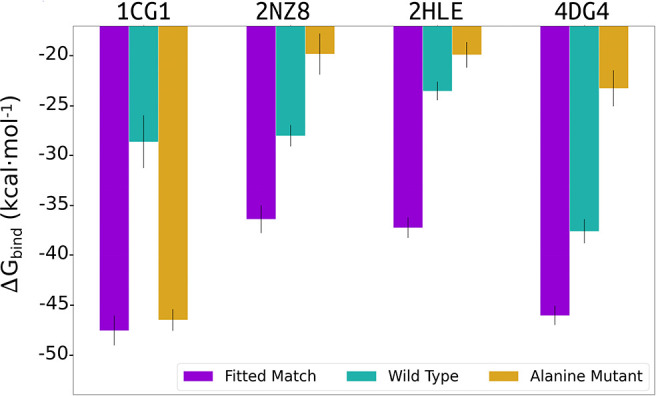
Rational design of specific inhibitors of protein-protein interactions is desirable for drug design to control cellular signal transduction but also for studying protein-protein interaction networks. We have developed a rapid computational approach to rationally design cyclic peptides that potentially bind at desired regions of the interface of protein-protein complexes. The methodology is based on comparing the protein backbone structure of short peptide segments (epitopes) at the protein-protein interface with a collection of cyclic peptide backbone structures. A cyclic peptide that matches the backbone structure of the segment is used as a template for a binder by adapting the amino acid side chains to the side chains found in the target complex. For a small library of cyclic peptides with known high resolution structures we found for the majority (~82%) of 154 protein-protein complexes at least one very well fitting match for a cyclic peptide template to a protein-protein interface segment. The majority of the constructed protein-cyclic peptide complexes was very stable during Molecular Dynamics simulations and showed an interaction energy score that was typically more favorable compared to interaction scores of typical peptide-protein complexes. Our cPEPmatch approach could be a promising approach for rapid suggestion of cyclic peptide binders that could be tested experimentally and further improved by chemical modification.
Keywords: cyclo peptide design; drug design with cyclo-peptides; protein binding modulation; protein interaction inhibition; protein-protein complexes; rational cyclo peptide binders.
Copyright © 2020 Santini and Zacharias.
Figures
Similar articles
-
Rapid Rational Design of Cyclic Peptides Mimicking Protein-Protein Interfaces.Methods Mol Biol. 2022;2405:231-244. doi: 10.1007/978-1-0716-1855-4_12. Methods Mol Biol. 2022. PMID: 35298817
-
De Novo Design of Cyclic Peptide Binders Based on Fragment Docking and Assembling.J Chem Inf Model. 2025 Apr 28;65(8):4206-4218. doi: 10.1021/acs.jcim.5c00088. Epub 2025 Apr 14. J Chem Inf Model. 2025. PMID: 40223692
-
The role of cation-pi interactions in biomolecular association. Design of peptides favoring interactions between cationic and aromatic amino acid side chains.J Am Chem Soc. 2001 Jul 4;123(26):6232-45. doi: 10.1021/ja010401u. J Am Chem Soc. 2001. PMID: 11427046
-
Computational Opportunities and Challenges in Finding Cyclic Peptide Modulators of Protein-Protein Interactions.Methods Mol Biol. 2019;2001:73-95. doi: 10.1007/978-1-4939-9504-2_5. Methods Mol Biol. 2019. PMID: 31134568 Review.
-
Crystal Structures of Protein-Bound Cyclic Peptides.Chem Rev. 2019 Sep 11;119(17):9861-9914. doi: 10.1021/acs.chemrev.8b00807. Epub 2019 May 2. Chem Rev. 2019. PMID: 31046237 Review.
Cited by
-
Heterocyclic Substitutions Greatly Improve Affinity and Stability of Folic Acid towards FRα. an In Silico Insight.Molecules. 2021 Feb 18;26(4):1079. doi: 10.3390/molecules26041079. Molecules. 2021. PMID: 33670773 Free PMC article.
-
What Makes a Good Protein-Protein Interaction Stabilizer: Analysis and Application of the Dual-Binding Mechanism.ACS Cent Sci. 2023 Apr 14;9(5):969-979. doi: 10.1021/acscentsci.3c00003. eCollection 2023 May 24. ACS Cent Sci. 2023. PMID: 37252344 Free PMC article.
-
Exploring a Structural Data Mining Approach to Design Linkers for Head-to-Tail Peptide Cyclization.J Chem Inf Model. 2023 Oct 23;63(20):6436-6450. doi: 10.1021/acs.jcim.3c00865. Epub 2023 Oct 12. J Chem Inf Model. 2023. PMID: 37827517 Free PMC article.
-
Cyclisation strategies for stabilising peptides with irregular conformations.RSC Med Chem. 2021 Apr 28;12(6):887-901. doi: 10.1039/d1md00098e. eCollection 2021 Jun 23. RSC Med Chem. 2021. PMID: 34263169 Free PMC article. Review.
-
Targeting the "undruggable" RAS with biologics.Adv Cancer Res. 2022;153:237-266. doi: 10.1016/bs.acr.2021.07.006. Epub 2021 Aug 13. Adv Cancer Res. 2022. PMID: 35101232 Free PMC article. Review.
References
-
- Chen F., Liu H., Sun H., Pan P., Li Y., Li D., et al. . (2016). Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 18, 22129–22139. 10.1039/C6CP03670H - DOI - PubMed
LinkOut - more resources
Full Text Sources