

Intricacies of aetiology in intrafamilial degenerative disease
- PMID: 33134917
- PMCID: PMC7585693
- DOI: 10.1093/braincomms/fcaa120
Intricacies of aetiology in intrafamilial degenerative disease
Abstract
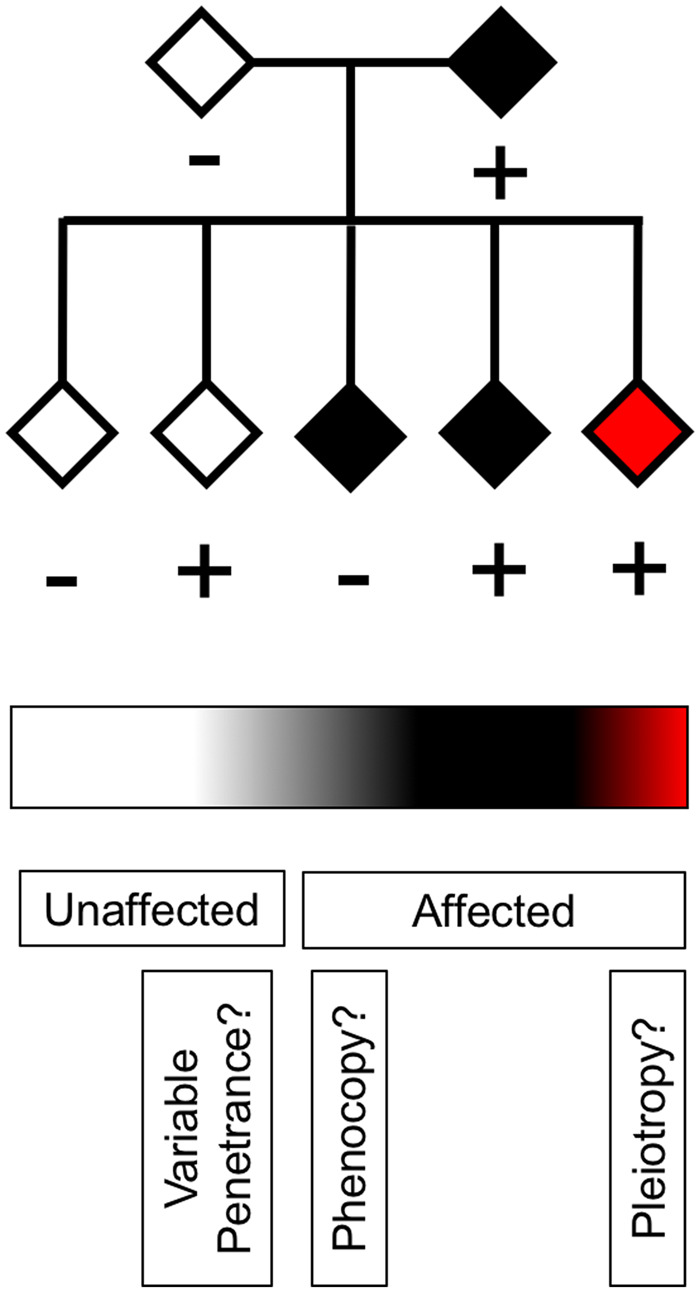
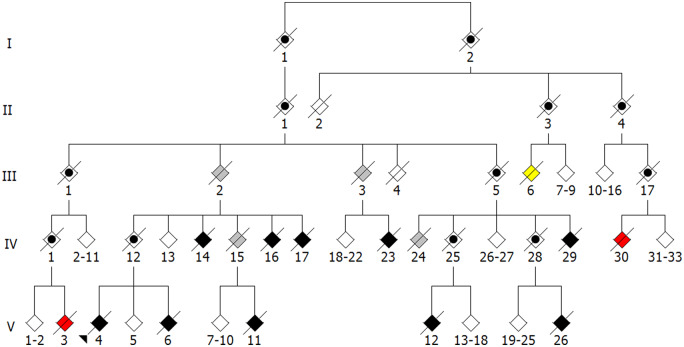
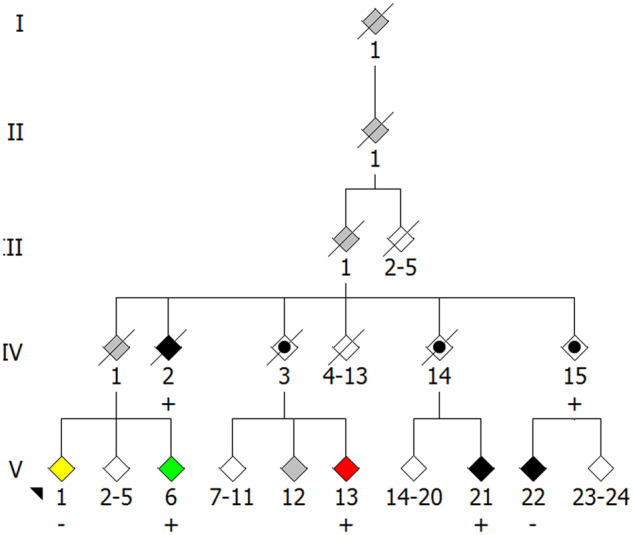
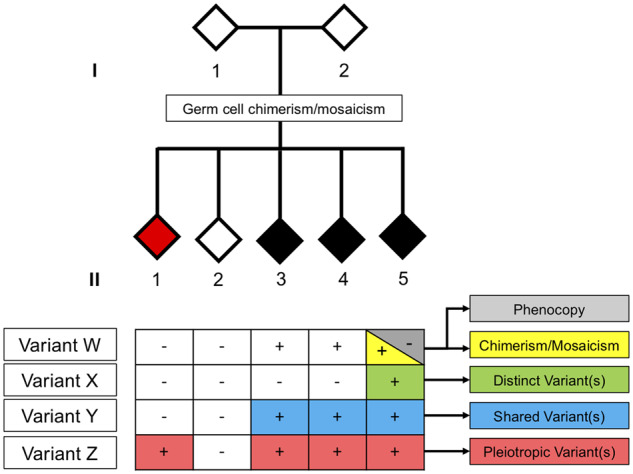
The genetic underpinnings of late-onset degenerative disease have typically been determined by screening families for the segregation of genetic variants with the disease trait in affected, but not unaffected, individuals. However, instances of intrafamilial etiological heterogeneity, where pathogenic variants in a culprit gene are not shared among all affected family members, continue to emerge and confound gene-discovery and genetic counselling efforts. Discordant intrafamilial cases lacking a mutation shared by other affected family members are described as disease phenocopies. This description often results in an over-simplified acceptance of an environmental cause of disease in the phenocopy cases, while the role of intrafamilial genetic heterogeneity, shared de novo mutations or epigenetic aberrations in such families is often ignored. On a related note, it is now evident that the same disease-associated variant can be present in individuals exhibiting clinically distinct phenotypes, thereby genetically uniting seemingly unrelated syndromes to form a spectrum of disease. Herein, we discuss the intricacies of determining complex degenerative disease aetiology and suggest alternative mechanisms of disease transmission that may account for the apparent missing heritability of disease.
Keywords: aetiology; inheritance; neurodegeneration; phenocopy; pleiotropy.
© The Author(s) (2020). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta 2015; 1852: 679–84. - PubMed
-
- Al-Chalabi A. Perspective: don't keep it in the family. Nature 2017; 550: S112. - PubMed
-
- Al-Chalabi A. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998; 7: 2045–50. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Medical