

Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses
- PMID: 33134918
- PMCID: PMC7585698
- DOI: 10.1093/braincomms/fcaa124
Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses
Abstract
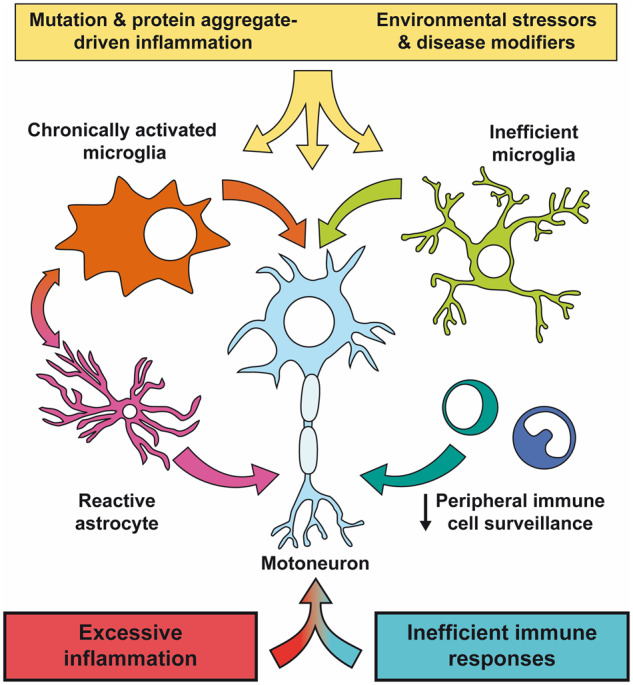
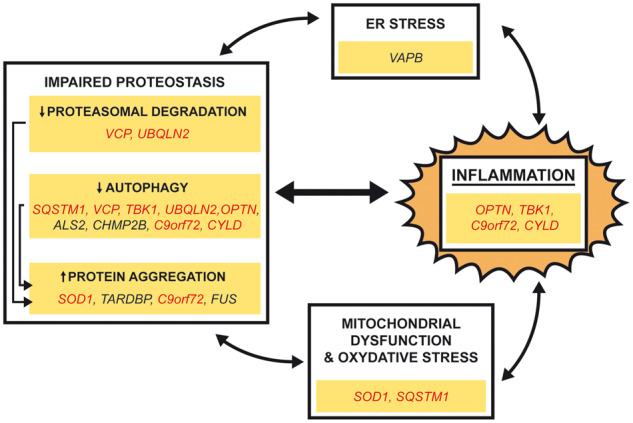
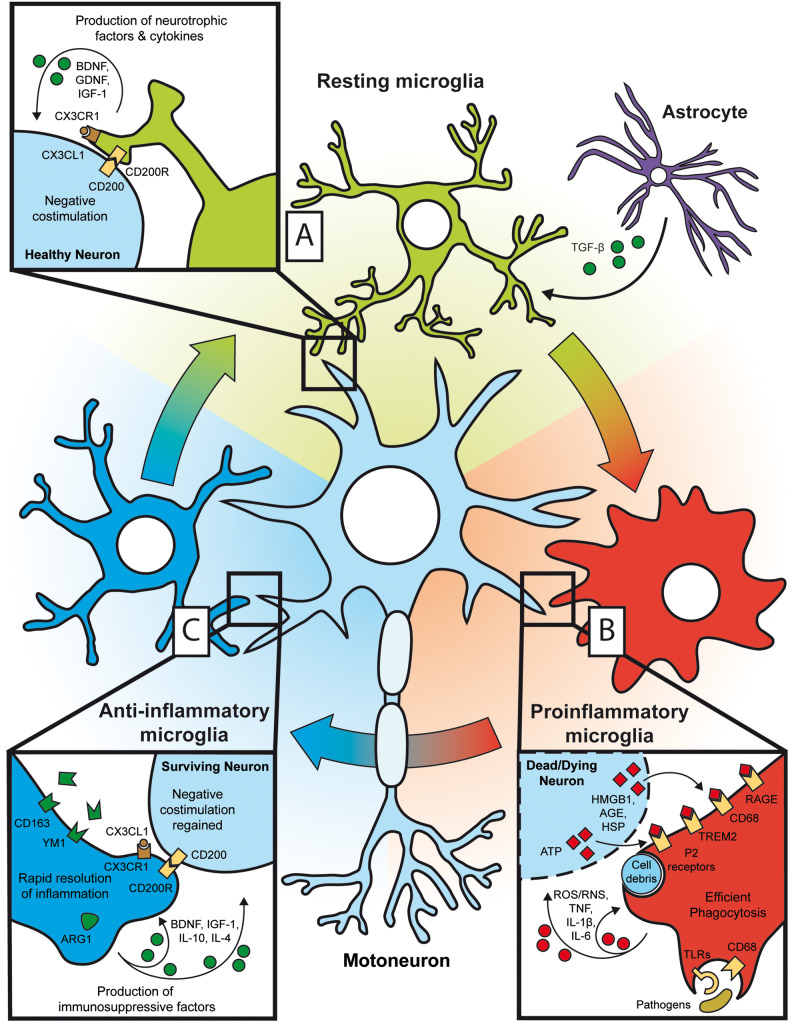
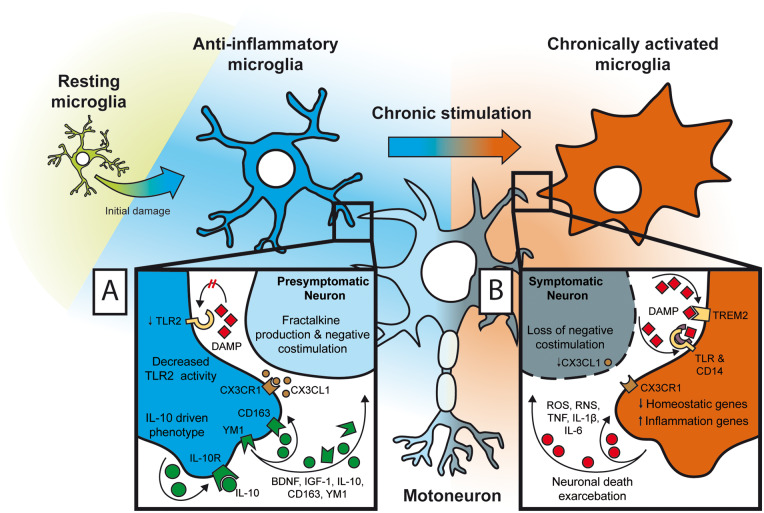
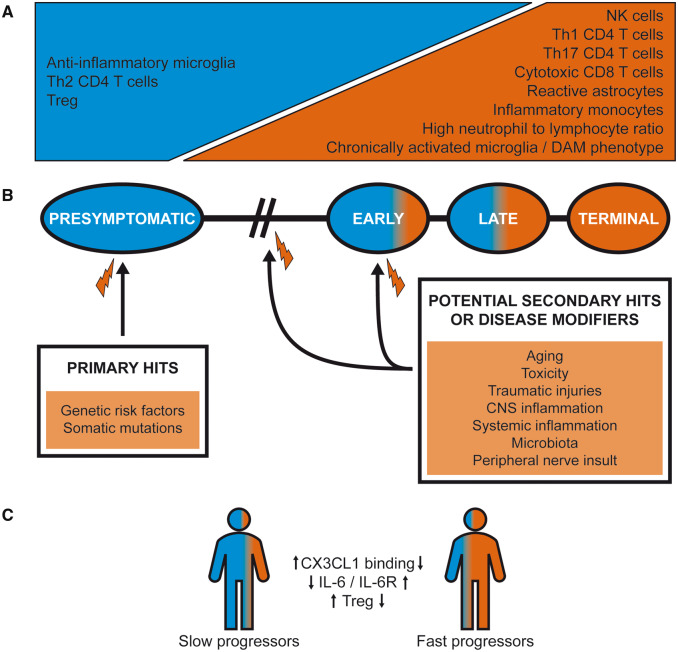
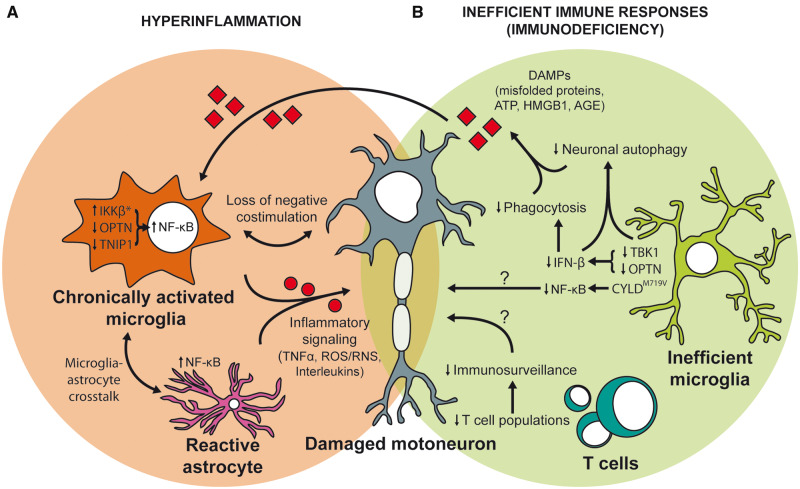
Despite wide genetic, environmental and clinical heterogeneity in amyotrophic lateral sclerosis, a rapidly fatal neurodegenerative disease targeting motoneurons, neuroinflammation is a common finding. It is marked by local glial activation, T cell infiltration and systemic immune system activation. The immune system has a prominent role in the pathogenesis of various chronic diseases, hence some of them, including some types of cancer, are successfully targeted by immunotherapeutic approaches. However, various anti-inflammatory or immunosuppressive therapies in amyotrophic lateral sclerosis have failed. This prompted increased scrutiny over the immune-mediated processes underlying amyotrophic lateral sclerosis. Perhaps the biggest conundrum is that amyotrophic lateral sclerosis pathogenesis exhibits features of three otherwise distinct immune dysfunctions-excessive inflammation, autoimmunity and inefficient immune responses. Epidemiological and genome-wide association studies show only minimal overlap between amyotrophic lateral sclerosis and autoimmune diseases, so excessive inflammation is usually thought to be secondary to protein aggregation, mitochondrial damage or other stresses. In contrast, several recently characterized amyotrophic lateral sclerosis-linked mutations, including those in TBK1, OPTN, CYLD and C9orf72, could lead to inefficient immune responses and/or damage pile-up, suggesting that an innate immunodeficiency may also be a trigger and/or modifier of this disease. In such cases, non-selective immunosuppression would further restrict neuroprotective immune responses. Here we discuss multiple layers of immune-mediated neuroprotection and neurotoxicity in amyotrophic lateral sclerosis. Particular focus is placed on individual patient mutations that directly or indirectly affect the immune system, and the mechanisms by which these mutations influence disease progression. The topic of immunity in amyotrophic lateral sclerosis is timely and relevant, because it is one of the few common and potentially malleable denominators in this heterogenous disease. Importantly, amyotrophic lateral sclerosis progression has recently been intricately linked to patient T cell and monocyte profiles, as well as polymorphisms in cytokine and chemokine receptors. For this reason, precise patient stratification based on immunophenotyping will be crucial for efficient therapies.
Keywords: amyotrophic lateral sclerosis; immunodeficiency; neuroimmunity neurodegeneration; neuroinflammation.
© The Author(s) (2020). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Similar articles
-
Inflammation in ALS/FTD pathogenesis.Acta Neuropathol. 2019 May;137(5):715-730. doi: 10.1007/s00401-018-1933-9. Epub 2018 Nov 21. Acta Neuropathol. 2019. PMID: 30465257 Free PMC article. Review.
-
Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis.J Neuroimmune Pharmacol. 2013 Sep;8(4):888-99. doi: 10.1007/s11481-013-9489-x. Epub 2013 Jul 25. J Neuroimmune Pharmacol. 2013. PMID: 23881705 Free PMC article. Review.
-
Neuroimmunity in amyotrophic lateral sclerosis: focus on microglia.Amyotroph Lateral Scler Frontotemporal Degener. 2020 May;21(3-4):159-166. doi: 10.1080/21678421.2019.1708949. Epub 2020 Jan 6. Amyotroph Lateral Scler Frontotemporal Degener. 2020. PMID: 31903792 Review.
-
Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia.J Mol Biol. 2019 Apr 19;431(9):1818-1829. doi: 10.1016/j.jmb.2019.02.004. Epub 2019 Feb 11. J Mol Biol. 2019. PMID: 30763568 Review.
-
Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact.Neurosci Biobehav Rev. 2021 Aug;127:958-978. doi: 10.1016/j.neubiorev.2021.06.027. Epub 2021 Jun 19. Neurosci Biobehav Rev. 2021. PMID: 34153344 Free PMC article. Review.
Cited by
-
Single-cell analysis reveals expanded CD8+ GZMK high T cells in CSF and shared peripheral clones in sporadic amyotrophic lateral sclerosis.Brain Commun. 2024 Nov 27;6(6):fcae428. doi: 10.1093/braincomms/fcae428. eCollection 2024. Brain Commun. 2024. PMID: 39659975 Free PMC article.
-
Identification and validation of a tear fluid-derived protein biomarker signature in patients with amyotrophic lateral sclerosis.Acta Neuropathol Commun. 2025 Sep 2;13(1):187. doi: 10.1186/s40478-025-02109-6. Acta Neuropathol Commun. 2025. PMID: 40898360 Free PMC article.
-
Aberrant protein aggregation in amyotrophic lateral sclerosis.J Neurol. 2024 Aug;271(8):4826-4851. doi: 10.1007/s00415-024-12485-z. Epub 2024 Jun 13. J Neurol. 2024. PMID: 38869826 Review.
-
Correlation between Retinal Vascularization and Disease Aggressiveness in Amyotrophic Lateral Sclerosis.Biomedicines. 2022 Sep 25;10(10):2390. doi: 10.3390/biomedicines10102390. Biomedicines. 2022. PMID: 36289652 Free PMC article.
-
Superoxide Dismutase-1 Intracellular Content in T Lymphocytes Associates with Increased Regulatory T Cell Level in Multiple Sclerosis Subjects Undergoing Immune-Modulating Treatment.Antioxidants (Basel). 2021 Dec 3;10(12):1940. doi: 10.3390/antiox10121940. Antioxidants (Basel). 2021. PMID: 34943042 Free PMC article.
References
-
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007; 10: 1538–43. - PubMed
-
- Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, et al. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem 2013; 126: 699–704. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous