

Cardiac fibrosis
- PMID: 33135058
- PMCID: PMC8152700
- DOI: 10.1093/cvr/cvaa324
Cardiac fibrosis
Abstract
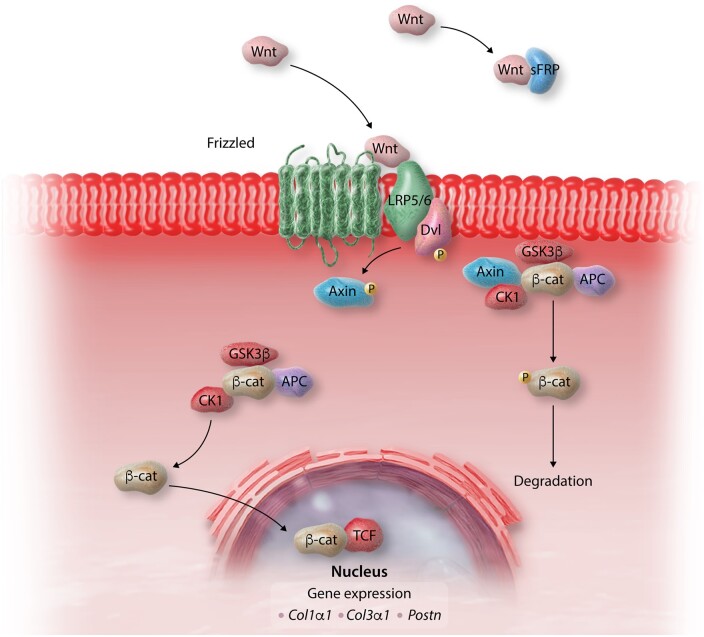
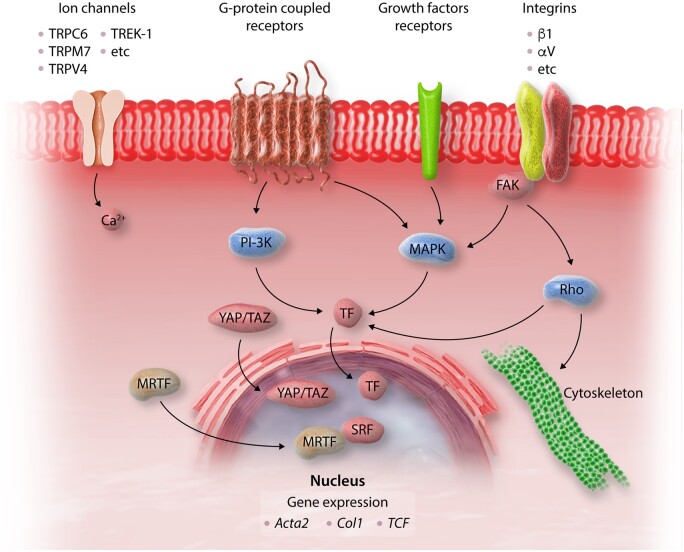
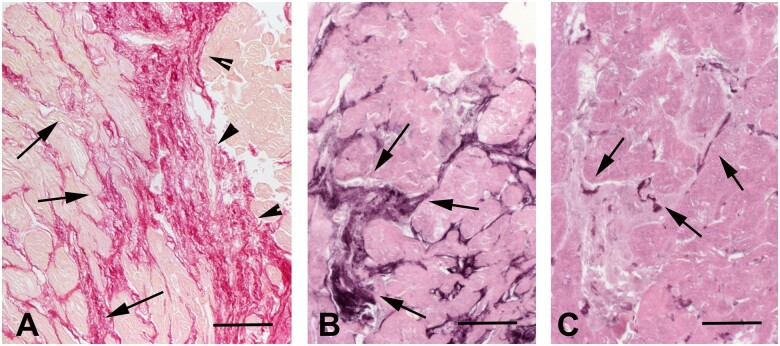
Myocardial fibrosis, the expansion of the cardiac interstitium through deposition of extracellular matrix proteins, is a common pathophysiologic companion of many different myocardial conditions. Fibrosis may reflect activation of reparative or maladaptive processes. Activated fibroblasts and myofibroblasts are the central cellular effectors in cardiac fibrosis, serving as the main source of matrix proteins. Immune cells, vascular cells and cardiomyocytes may also acquire a fibrogenic phenotype under conditions of stress, activating fibroblast populations. Fibrogenic growth factors (such as transforming growth factor-β and platelet-derived growth factors), cytokines [including tumour necrosis factor-α, interleukin (IL)-1, IL-6, IL-10, and IL-4], and neurohumoral pathways trigger fibrogenic signalling cascades through binding to surface receptors, and activation of downstream signalling cascades. In addition, matricellular macromolecules are deposited in the remodelling myocardium and regulate matrix assembly, while modulating signal transduction cascades and protease or growth factor activity. Cardiac fibroblasts can also sense mechanical stress through mechanosensitive receptors, ion channels and integrins, activating intracellular fibrogenic cascades that contribute to fibrosis in response to pressure overload. Although subpopulations of fibroblast-like cells may exert important protective actions in both reparative and interstitial/perivascular fibrosis, ultimately fibrotic changes perturb systolic and diastolic function, and may play an important role in the pathogenesis of arrhythmias. This review article discusses the molecular mechanisms involved in the pathogenesis of cardiac fibrosis in various myocardial diseases, including myocardial infarction, heart failure with reduced or preserved ejection fraction, genetic cardiomyopathies, and diabetic heart disease. Development of fibrosis-targeting therapies for patients with myocardial diseases will require not only understanding of the functional pluralism of cardiac fibroblasts and dissection of the molecular basis for fibrotic remodelling, but also appreciation of the pathophysiologic heterogeneity of fibrosis-associated myocardial disease.
Keywords: Extracellular matrix; Fibrosis; Growth factor; Heart failure; Myofibroblast.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2020. For permissions, please email: journals.permissions@oup.com.
Figures
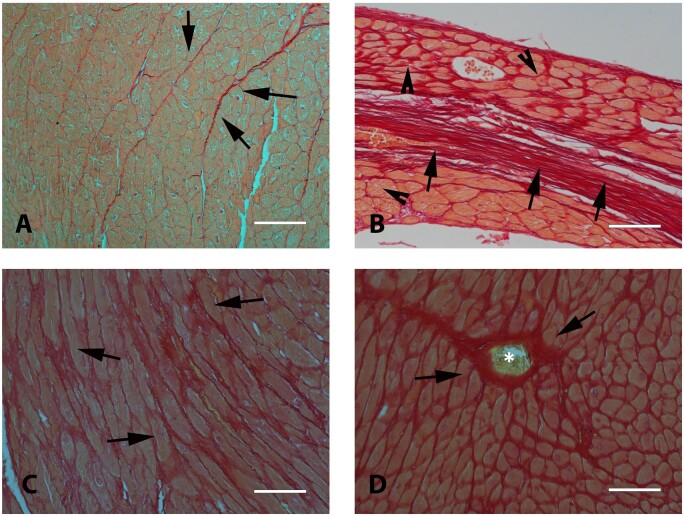
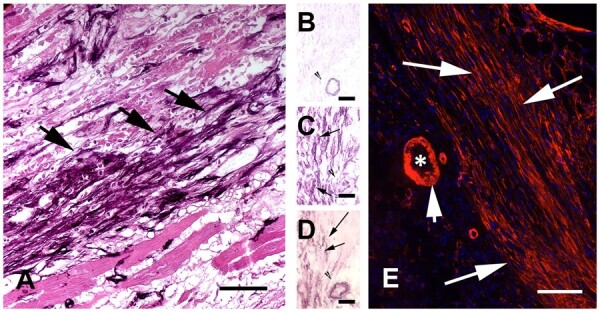
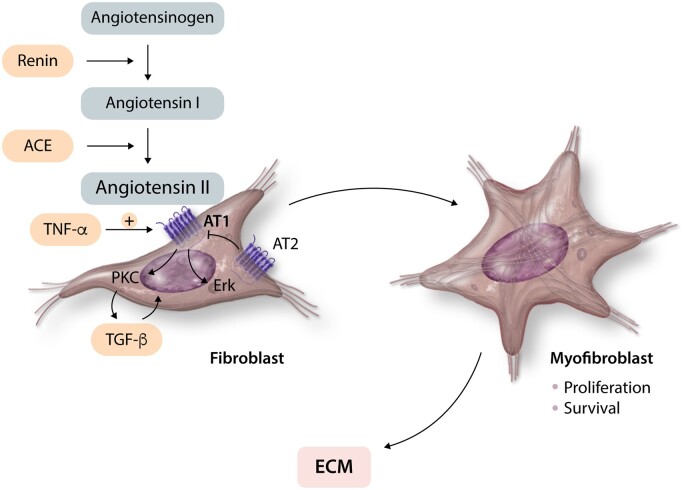
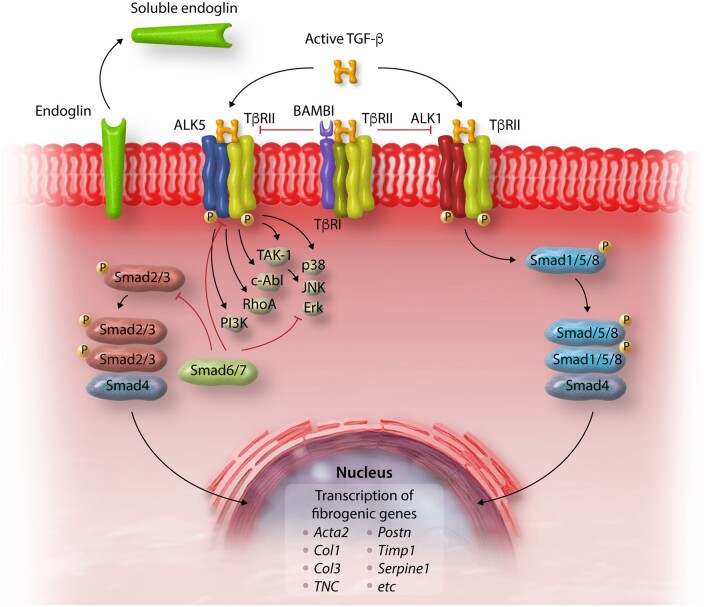
References
-
- Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 2019;65:70–99. - PubMed
-
- Gao XM, White DA, Dart AM, Du XJ.. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 2012;134:156–179. - PubMed
-
- Borg TK, Caulfield JB.. The collagen matrix of the heart. Fed Proc 1981;40:2037–2041. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
