

Inferring TF activities and activity regulators from gene expression data with constraints from TF perturbation data
- PMID: 33135076
- PMCID: PMC8189679
- DOI: 10.1093/bioinformatics/btaa947
Inferring TF activities and activity regulators from gene expression data with constraints from TF perturbation data
Abstract
Motivation: The activity of a transcription factor (TF) in a sample of cells is the extent to which it is exerting its regulatory potential. Many methods of inferring TF activity from gene expression data have been described, but due to the lack of appropriate large-scale datasets, systematic and objective validation has not been possible until now.
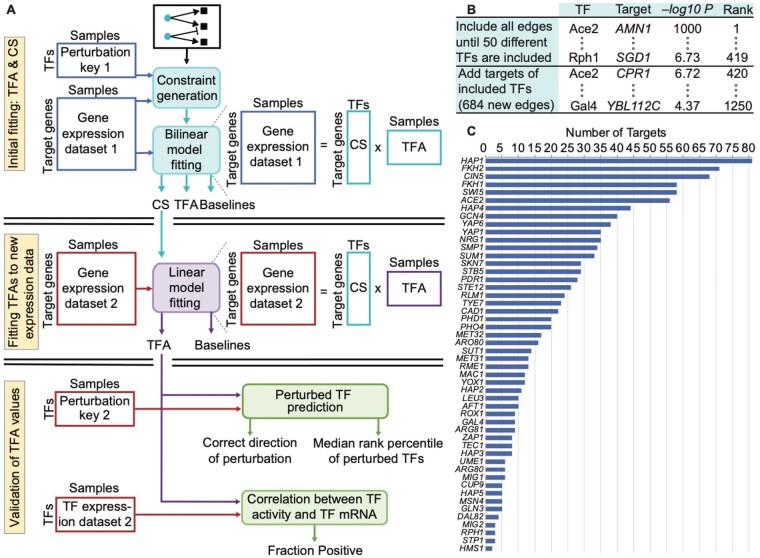
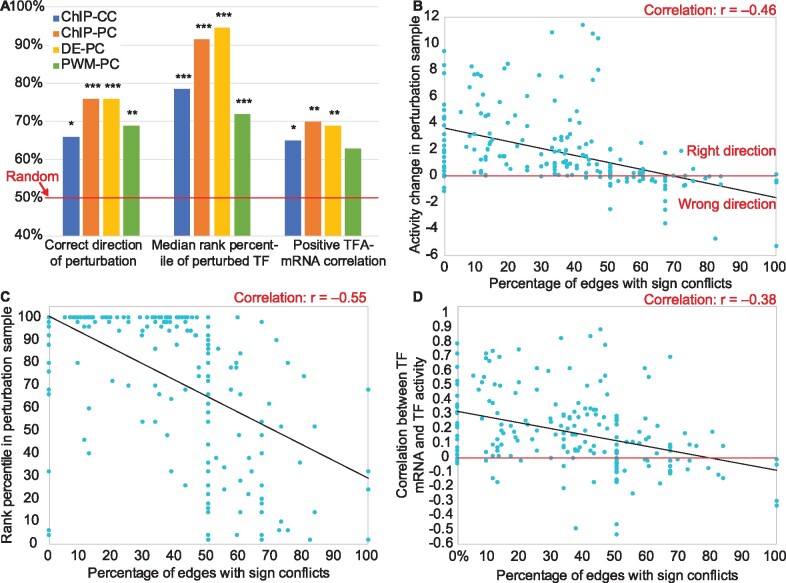
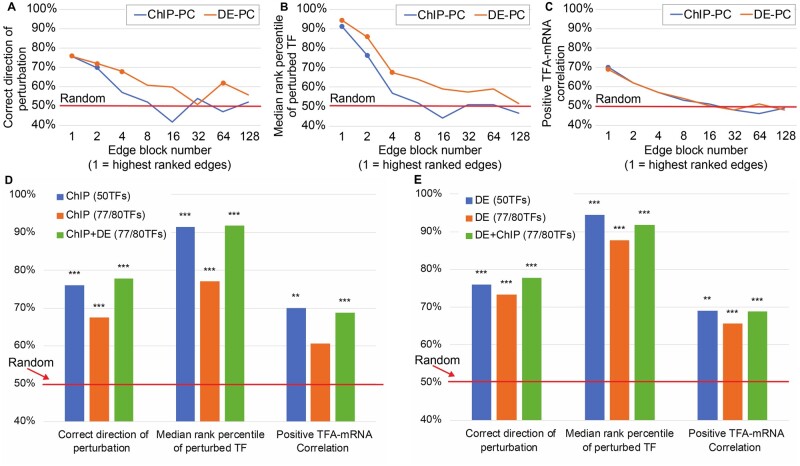
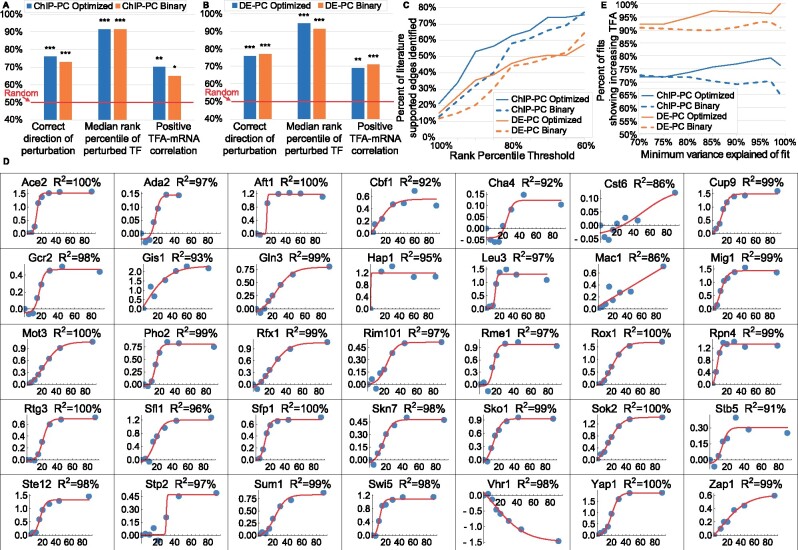
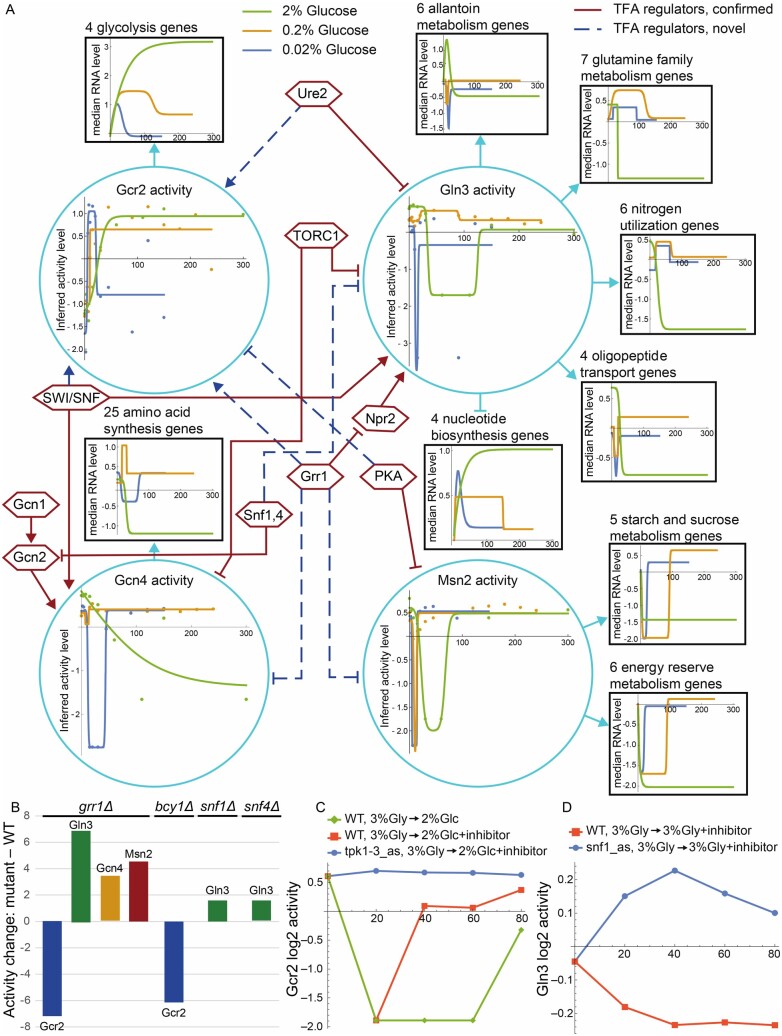
Results: We systematically evaluate and optimize the approach to TF activity inference in which a gene expression matrix is factored into a condition-independent matrix of control strengths and a condition-dependent matrix of TF activity levels. We find that expression data in which the activities of individual TFs have been perturbed are both necessary and sufficient for obtaining good performance. To a considerable extent, control strengths inferred using expression data from one growth condition carry over to other conditions, so the control strength matrices derived here can be used by others. Finally, we apply these methods to gain insight into the upstream factors that regulate the activities of yeast TFs Gcr2, Gln3, Gcn4 and Msn2.
Availability and implementation: Evaluation code and data are available at https://doi.org/10.5281/zenodo.4050573.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press.
Figures
Similar articles
-
Inferring condition-specific modulation of transcription factor activity in yeast through regulon-based analysis of genomewide expression.PLoS One. 2008 Sep 3;3(9):e3112. doi: 10.1371/journal.pone.0003112. PLoS One. 2008. PMID: 18769540 Free PMC article.
-
De novo motif discovery facilitates identification of interactions between transcription factors in Saccharomyces cerevisiae.Bioinformatics. 2012 Mar 1;28(5):701-8. doi: 10.1093/bioinformatics/bts002. Epub 2012 Jan 11. Bioinformatics. 2012. PMID: 22238267
-
TF-centered downstream gene set enrichment analysis: Inference of causal regulators by integrating TF-DNA interactions and protein post-translational modifications information.BMC Bioinformatics. 2010 Dec 14;11 Suppl 11(Suppl 11):S5. doi: 10.1186/1471-2105-11-S11-S5. BMC Bioinformatics. 2010. PMID: 21172055 Free PMC article.
-
[Alternative ways of stress regulation in cells of Saccharomyces cerevisiae: transcriptional activators Msn2 and Msn4].Tsitologiia. 2009;51(3):271-8. Tsitologiia. 2009. PMID: 19435282 Review. Russian.
-
Evolutionary Transition of GAL Regulatory Circuit from Generalist to Specialist Function in Ascomycetes.Trends Microbiol. 2018 Aug;26(8):692-702. doi: 10.1016/j.tim.2017.12.008. Trends Microbiol. 2018. PMID: 29395731 Review.
Cited by
-
GOAT: Gene-level biomarker discovery from multi-Omics data using graph ATtention neural network for eosinophilic asthma subtype.Bioinformatics. 2023 Oct 3;39(10):btad582. doi: 10.1093/bioinformatics/btad582. Bioinformatics. 2023. PMID: 37740295 Free PMC article.
-
Identifying strengths and weaknesses of methods for computational network inference from single-cell RNA-seq data.G3 (Bethesda). 2023 Mar 9;13(3):jkad004. doi: 10.1093/g3journal/jkad004. G3 (Bethesda). 2023. PMID: 36626328 Free PMC article.
-
Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling.Genes (Basel). 2023 Jun 10;14(6):1245. doi: 10.3390/genes14061245. Genes (Basel). 2023. PMID: 37372428 Free PMC article.
-
Prostate cancers with distinct transcriptional programs in Black and White men.Genome Med. 2024 Jul 23;16(1):92. doi: 10.1186/s13073-024-01361-0. Genome Med. 2024. PMID: 39044302 Free PMC article.
-
Integrated analysis of ovarian cancer patients from prospective transcription factor activity reveals subtypes of prognostic significance.Heliyon. 2023 May 11;9(5):e16147. doi: 10.1016/j.heliyon.2023.e16147. eCollection 2023 May. Heliyon. 2023. PMID: 37215759 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
