

Synthetic Lethality through the Lens of Medicinal Chemistry
- PMID: 33135887
- PMCID: PMC8015234
- DOI: 10.1021/acs.jmedchem.0c00766
Synthetic Lethality through the Lens of Medicinal Chemistry
Abstract
Personalized medicine and therapies represent the goal of modern medicine, as drug discovery strives to move away from one-cure-for-all and makes use of the various targets and biomarkers within differing disease areas. This approach, especially in oncology, is often undermined when the cells make use of alternative survival pathways. As such, acquired resistance is unfortunately common. In order to combat this phenomenon, synthetic lethality is being investigated, making use of existing genetic fragilities within the cancer cell. This Perspective highlights exciting targets within synthetic lethality, (PARP, ATR, ATM, DNA-PKcs, WEE1, CDK12, RAD51, RAD52, and PD-1) and discusses the medicinal chemistry programs being used to interrogate them, the challenges these programs face, and what the future holds for this promising field.
Conflict of interest statement
The authors declare no competing financial interest.
Figures




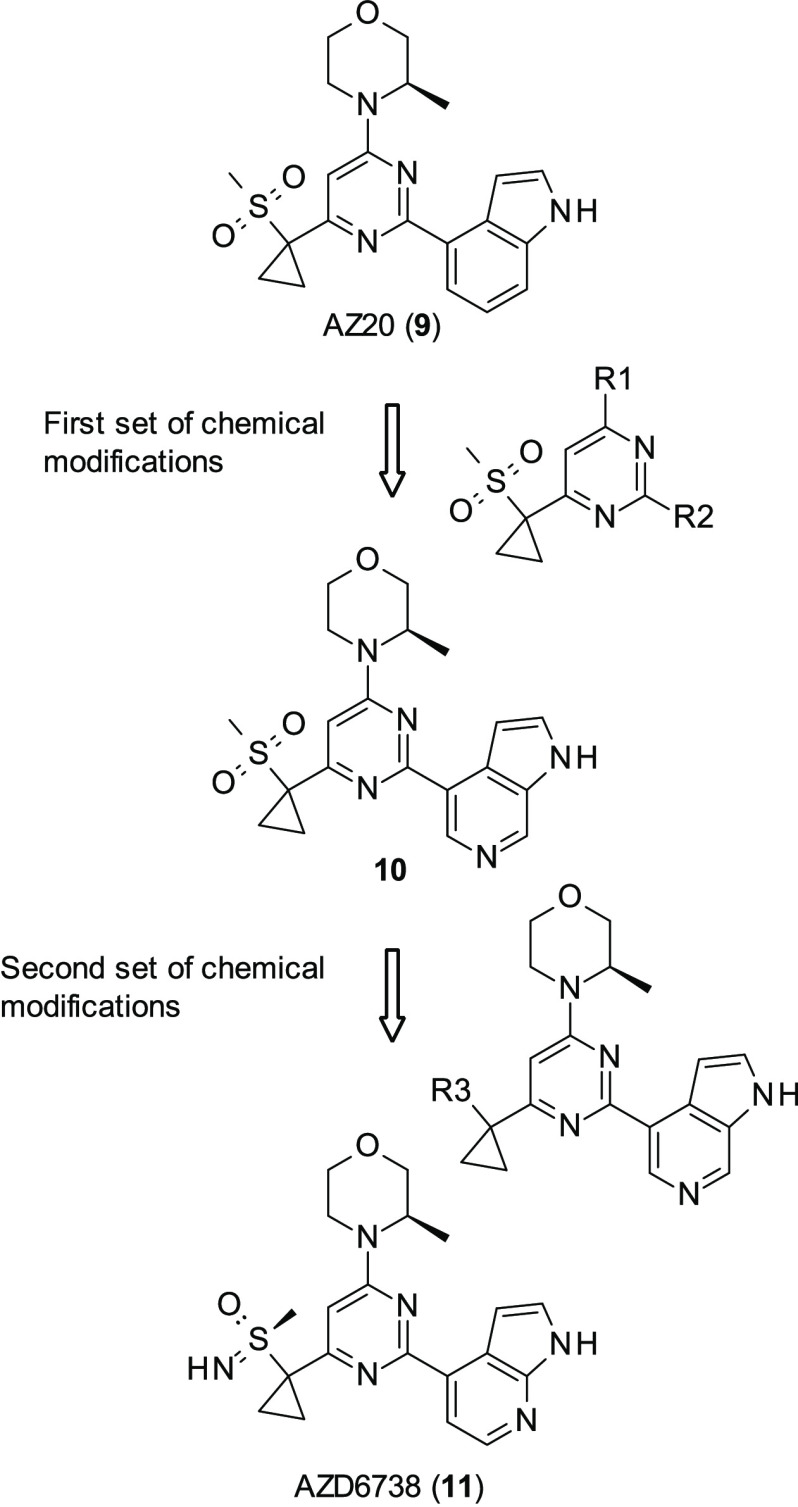






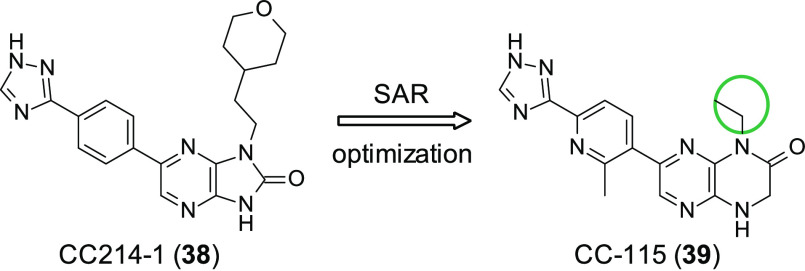





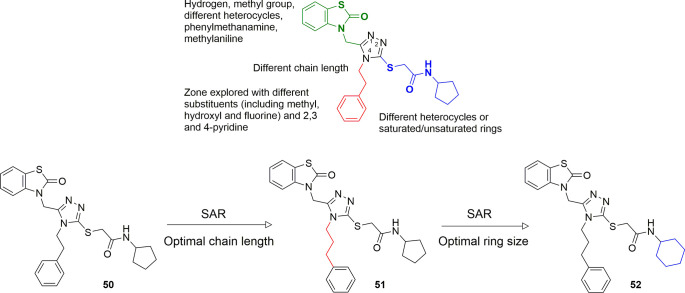






References
-
- Hochhaus A.; Larson R. A.; Guilhot F.; Radich J. P.; Branford S.; Hughes T. P.; Baccarani M.; Deininger M. W.; Cervantes F.; Fujihara S.; Ortmann C. E.; Menssen H. D.; Kantarjian H.; O’Brien S. G.; Druker B. J. Long-Term outcomes of Imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med. 2017, 376 (10), 917–927. 10.1056/NEJMoa1609324. - DOI - PMC - PubMed
-
- Chapman P. B.; Hauschild A.; Robert C.; Haanen J. B.; Ascierto P.; Larkin J.; Dummer R.; Garbe C.; Testori A.; Maio M.; Hogg D.; Lorigan P.; Lebbe C.; Jouary T.; Schadendorf D.; Ribas A.; O’Day S. J.; Sosman J. A.; Kirkwood J. M.; Eggermont A. M.; Dreno B.; Nolop K.; Li J.; Nelson B.; Hou J.; Lee R. J.; Flaherty K. T.; McArthur G. A. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364 (26), 2507–2516. 10.1056/NEJMoa1103782. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Chemical Information
Research Materials
Miscellaneous
