

Many, but not all, lineage-specific genes can be explained by homology detection failure
- PMID: 33137085
- PMCID: PMC7660931
- DOI: 10.1371/journal.pbio.3000862
Many, but not all, lineage-specific genes can be explained by homology detection failure
Abstract
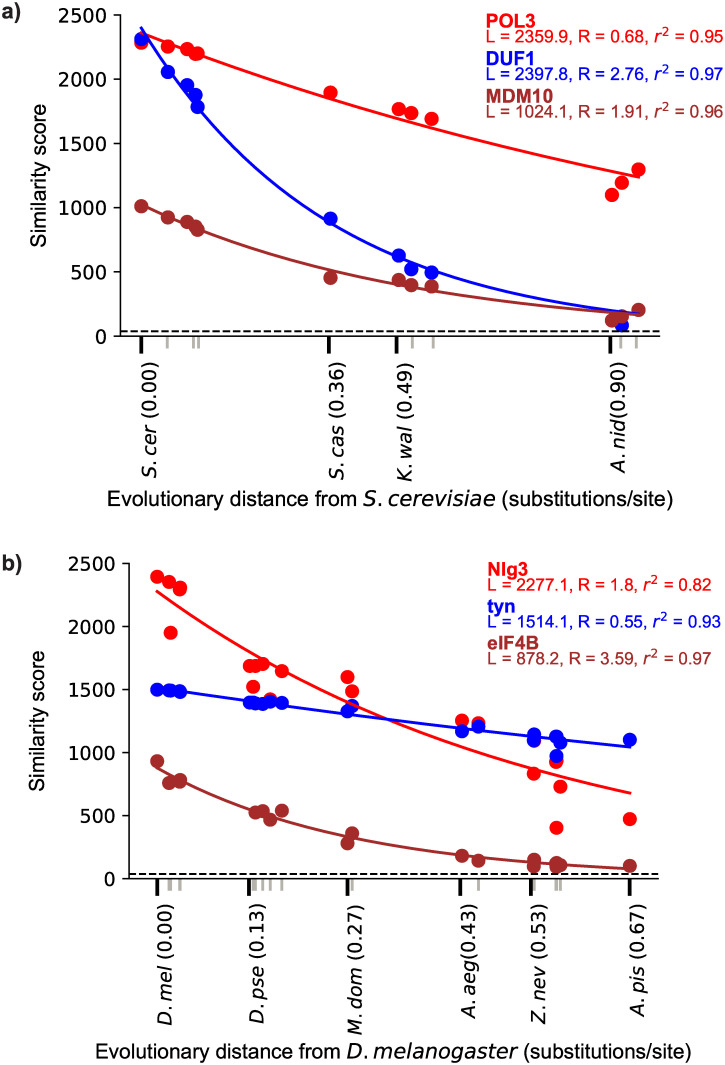
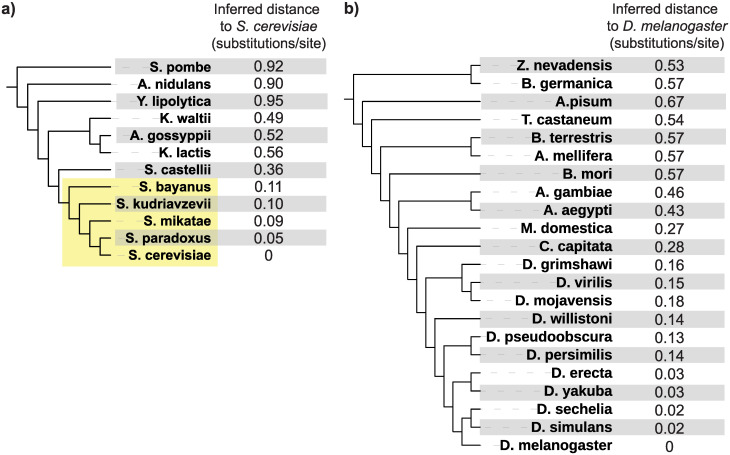
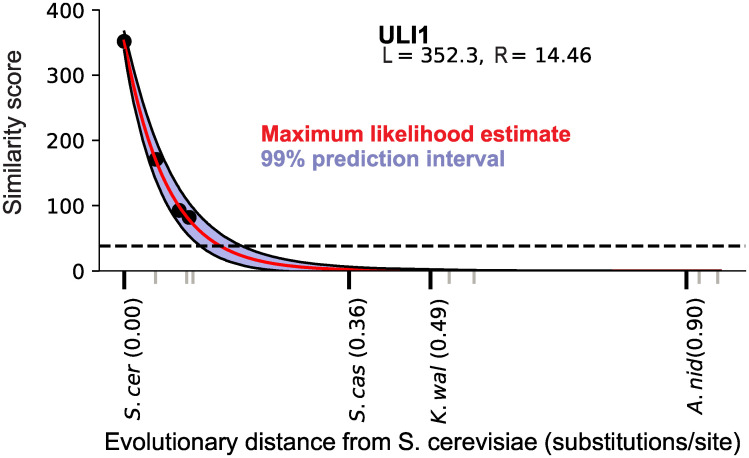
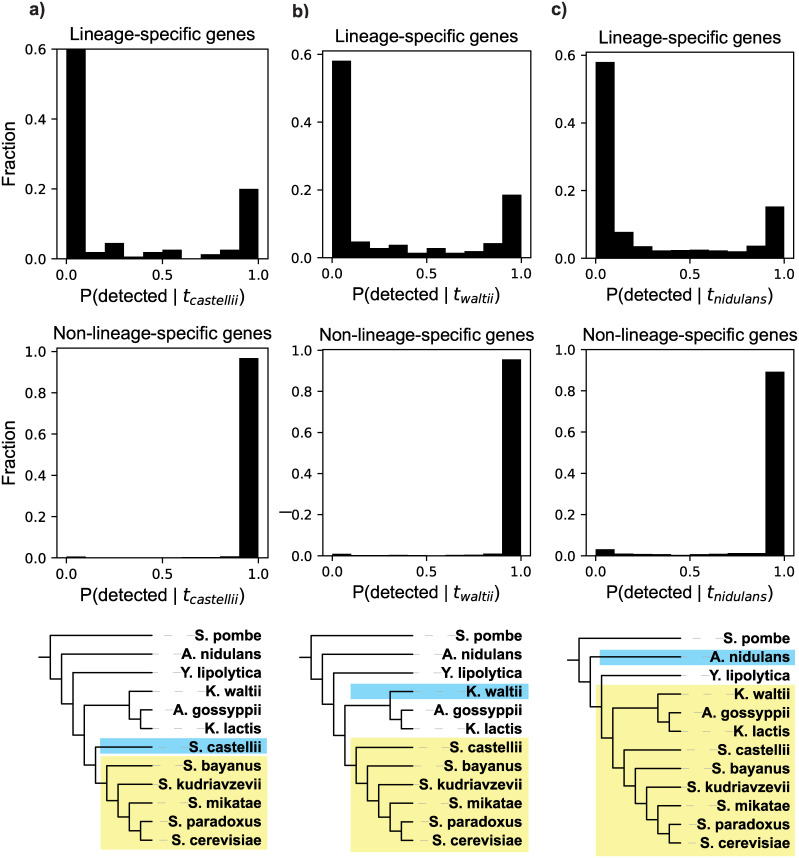
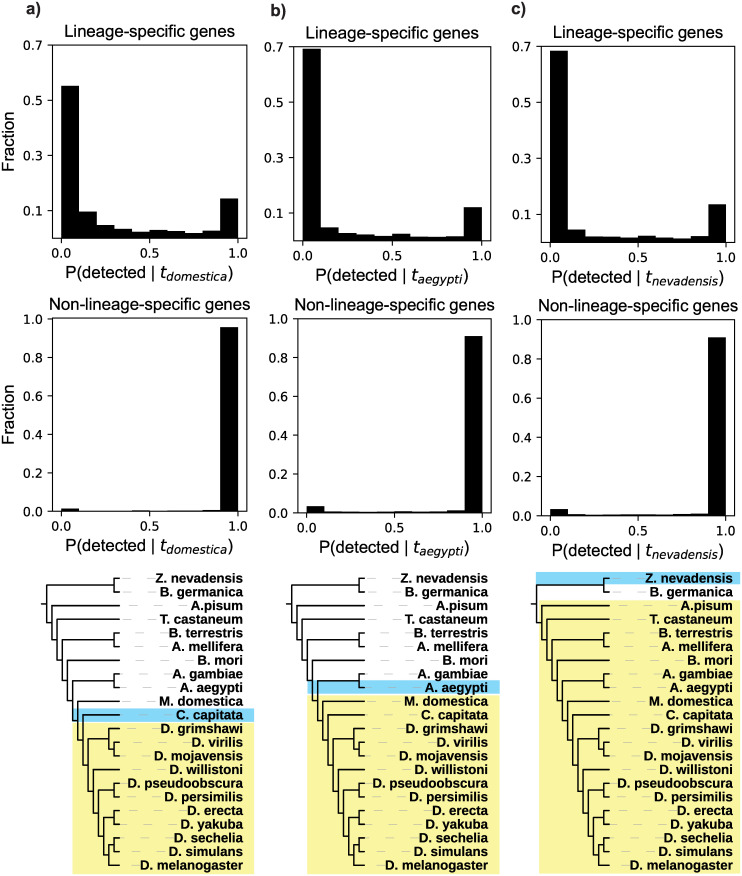
Genes for which homologs can be detected only in a limited group of evolutionarily related species, called "lineage-specific genes," are pervasive: Essentially every lineage has them, and they often comprise a sizable fraction of the group's total genes. Lineage-specific genes are often interpreted as "novel" genes, representing genetic novelty born anew within that lineage. Here, we develop a simple method to test an alternative null hypothesis: that lineage-specific genes do have homologs outside of the lineage that, even while evolving at a constant rate in a novelty-free manner, have merely become undetectable by search algorithms used to infer homology. We show that this null hypothesis is sufficient to explain the lack of detected homologs of a large number of lineage-specific genes in fungi and insects. However, we also find that a minority of lineage-specific genes in both clades are not well explained by this novelty-free model. The method provides a simple way of identifying which lineage-specific genes call for special explanations beyond homology detection failure, highlighting them as interesting candidates for further study.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
