

RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status
- PMID: 33138032
- PMCID: PMC7693555
- DOI: 10.3390/cancers12113178
RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status
Abstract
PTEN mutation occurs in a variety of aggressive cancers and is associated with poor patient outcomes. Recent studies have linked mutational loss of PTEN to reduced RAD51 expression and function, a key factor involved in the homologous recombination (HR) pathway. However, these studies remain controversial, as they fail to establish a definitive causal link to RAD51 expression that is PTEN-dependent, while other studies have not been able to recapitulate the relationship between the PTEN expression and the RAD51/HR function. Resolution of this apparent conundrum is essential due to the clinically-significant implication that PTEN-deficient tumors may be sensitive to poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) commonly used in the clinical management of BRCA-mutated and other HR-deficient (HRD) tumors.
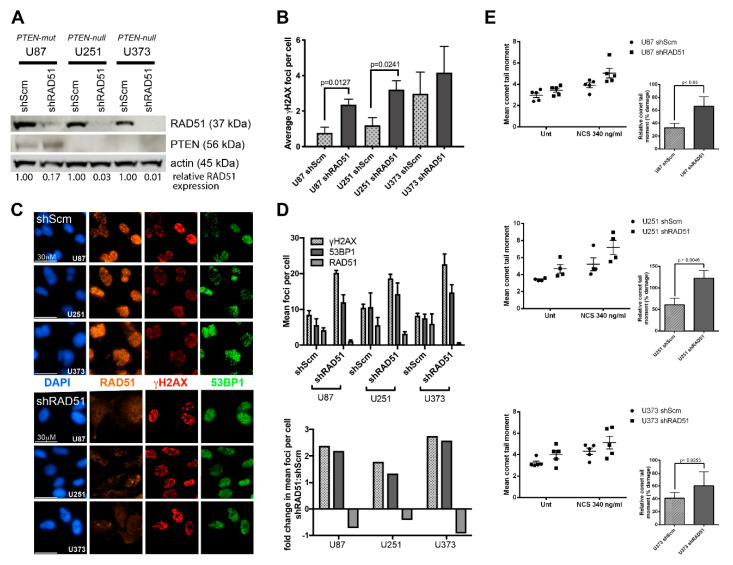
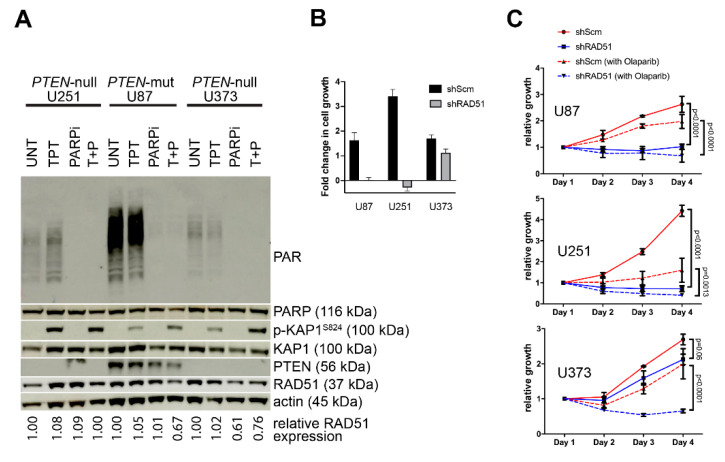
Methods: Primary Pten-deficient (and corresponding wild-type) mouse embryonic fibroblasts (MEFs) and astrocytes and PTEN-null human tumor cell lines and primary cells were assessed for RAD51 expression (via the Western blot analysis) and DNA damage repair analyses (via alkali comet and γH2AX foci assays). RAD51 foci analysis was used to measure HR-dependent DNA repair. Xrcc2-deficient MEFs served as an HR-deficient control, while the stable knockdown of RAD51 (shRAD51) served to control for the relative RAD51/HR-mediated repair and the phospho-53BP1 foci analysis served to confirm and measure non-homologous end joining (NHEJ) activity in PTEN-deficient and shRAD51-expressing (HRD) lines. Cell proliferation studies were used to measure any potential added sensitivity of PTEN-null cells to the clinically-relevant PARPi, olaparib. RAD51 levels and DNA damage response signaling were assessed in PTEN-mutant brain tumor initiating cells (BTICs) derived from primary and recurrent glioblastoma multiforme (GBM) patients, while expression of RAD51 and its paralogs were examined as a function of the PTEN status in the RNA expression datasets isolated from primary GBM tumor specimens and BTICs.
Results: Pten knockout primary murine cells display unaltered RAD51 expression, endogenous and DNA strand break-induced RAD51 foci and robust DNA repair activity. Defective HR was only observed in the cells lacking Xrcc2. Likewise, human glioblastoma multiforme (GBM) cell lines with known PTEN deficiency (U87, PTEN-mutated; U251 and U373, PTEN-null) show apparent expression of RAD51 and display efficient DNA repair activity. Only GBM cells stably expressing shRNAs against RAD51 (shRAD51) display dysfunctional DNA repair activity and reduced proliferative capacity, which is exacerbated by PARPi treatment. Furthermore, GBM patient-derived BTICs displayed robust RAD51 expression and intact DNA damage response signaling in spite of PTEN-inactivating mutations. RNA expression analysis of primary GBM tissue specimens and BTICs demonstrate stable levels of RAD51 and its paralogs (RAD51B, RAD51C, RAD51D, XRCC2, XRCC3, and DMC1), regardless of the PTEN mutational status.
Conclusions: Our findings demonstrate definitively that PTEN loss does not alter the RAD51 expression, its paralogs, or the HR activity. Furthermore, deficiency in PTEN alone is not sufficient to impart enhanced sensitivity to PARPi associated with HRD. This study is the first to unequivocally demonstrate that PTEN deficiency is not linked to the RAD51 expression or the HR activity amongst primary neural and non-neural Pten-null cells, PTEN-deficient tumor cell lines, and primary PTEN-mutant GBM patient-derived tissue specimens and BTICs.
Keywords: DNA damage; HRD; PARP inhibitor; PTEN; RAD51; RAD51 foci; RNA expression; alkaline comet assay; base excision repair; brain tumor initiating cells; combination therapy; glioblastoma multiforme; homologous recombination; olaparib; synthetic lethality; γH2AX foci.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Tousled-like kinase loss confers PARP inhibitor resistance in BRCA1-mutated cancers by impeding non-homologous end joining repair.Mol Med. 2025 Jan 22;31(1):18. doi: 10.1186/s10020-025-01066-z. Mol Med. 2025. PMID: 39844055 Free PMC article.
-
Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells.BMC Cancer. 2014 Mar 13;14:179. doi: 10.1186/1471-2407-14-179. BMC Cancer. 2014. PMID: 24625059 Free PMC article.
-
Homologous recombination deficiency real-time clinical assays, ready or not?Gynecol Oncol. 2020 Dec;159(3):877-886. doi: 10.1016/j.ygyno.2020.08.035. Epub 2020 Sep 20. Gynecol Oncol. 2020. PMID: 32967790 Review.
-
PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors.Cancer Res. 2010 Jul 1;70(13):5457-64. doi: 10.1158/0008-5472.CAN-09-4295. Epub 2010 Jun 8. Cancer Res. 2010. PMID: 20530668 Free PMC article.
-
Cracking the homologous recombination deficiency code: how to identify responders to PARP inhibitors.Eur J Cancer. 2022 May;166:87-99. doi: 10.1016/j.ejca.2022.01.037. Epub 2022 Mar 10. Eur J Cancer. 2022. PMID: 35279473 Review.
Cited by
-
Homologous recombination, cancer and the 'RAD51 paradox'.NAR Cancer. 2021 May 17;3(2):zcab016. doi: 10.1093/narcan/zcab016. eCollection 2021 Jun. NAR Cancer. 2021. PMID: 34316706 Free PMC article.
-
PTEN: an emerging target in rheumatoid arthritis?Cell Commun Signal. 2024 Apr 26;22(1):246. doi: 10.1186/s12964-024-01618-6. Cell Commun Signal. 2024. PMID: 38671436 Free PMC article. Review.
-
Mechanisms driving the immunoregulatory function of cancer cells.Nat Rev Cancer. 2023 Apr;23(4):193-215. doi: 10.1038/s41568-022-00544-4. Epub 2023 Jan 30. Nat Rev Cancer. 2023. PMID: 36717668 Review.
-
PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination.Int J Mol Sci. 2022 Oct 25;23(21):12876. doi: 10.3390/ijms232112876. Int J Mol Sci. 2022. PMID: 36361678 Free PMC article.
-
Circulating Tumor Cells in Breast Cancer Patients: A Balancing Act between Stemness, EMT Features and DNA Damage Responses.Cancers (Basel). 2022 Feb 16;14(4):997. doi: 10.3390/cancers14040997. Cancers (Basel). 2022. PMID: 35205744 Free PMC article. Review.
References
-
- Steck P.A., Pershouse M.A., Jasser S.A., Yung W.K., Lin H., Ligon A.H., Langford L.A., Baumgard M.L., Hattier T., Davis T., et al. Identification of a Candidate Tumour Suppressor Gene, MMAC1, At Chromosome 10q23.3 That Is Mutated in Multiple Advanced Cancers. Nat. Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
