

Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19
- PMID: 33138195
- PMCID: PMC7693119
- DOI: 10.3390/cells9112374
Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19
Abstract
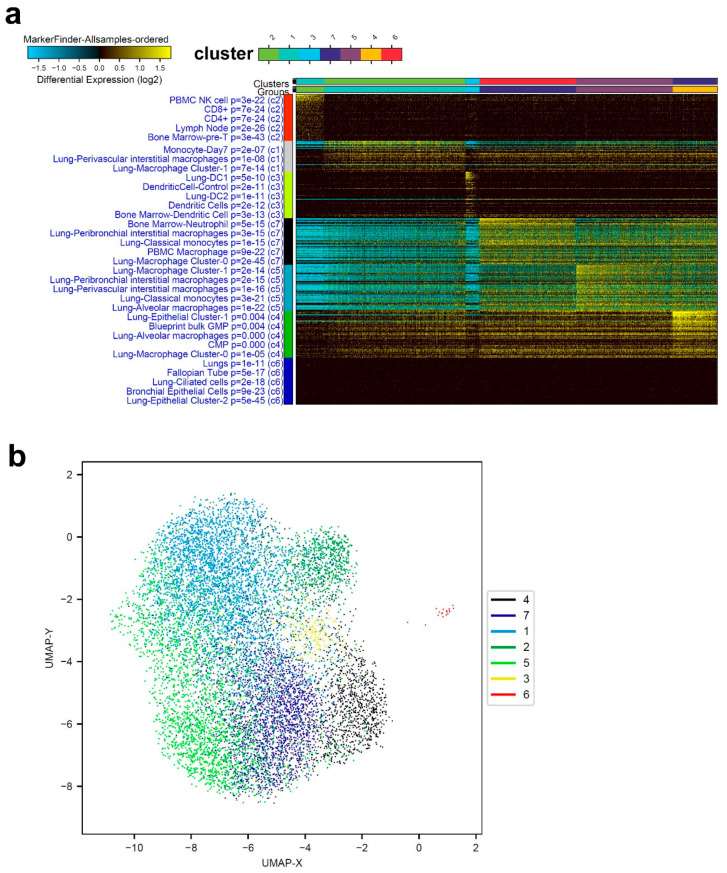
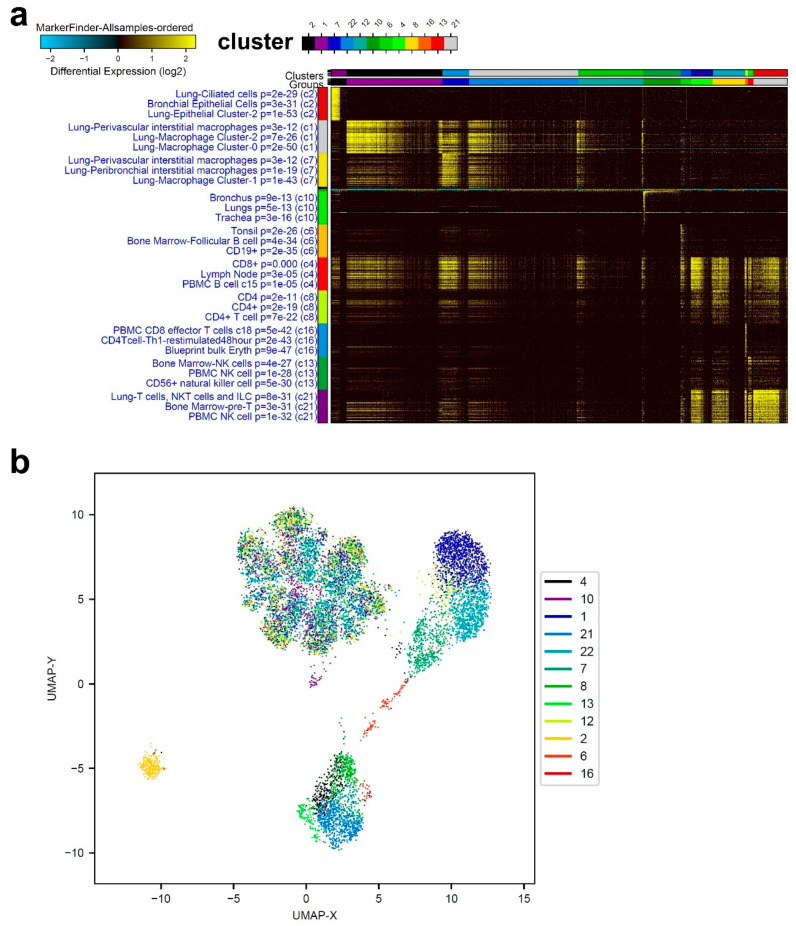
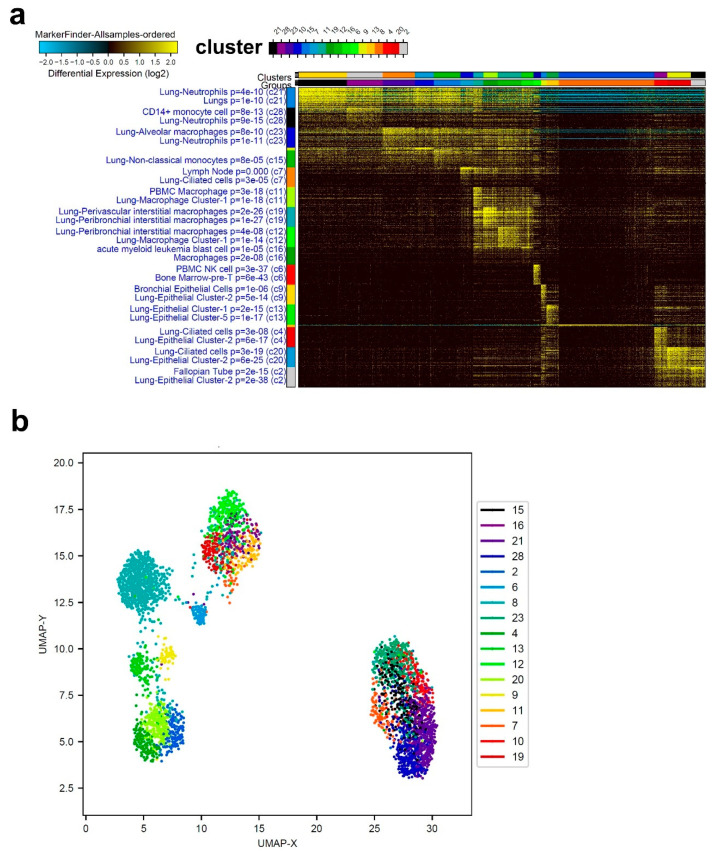
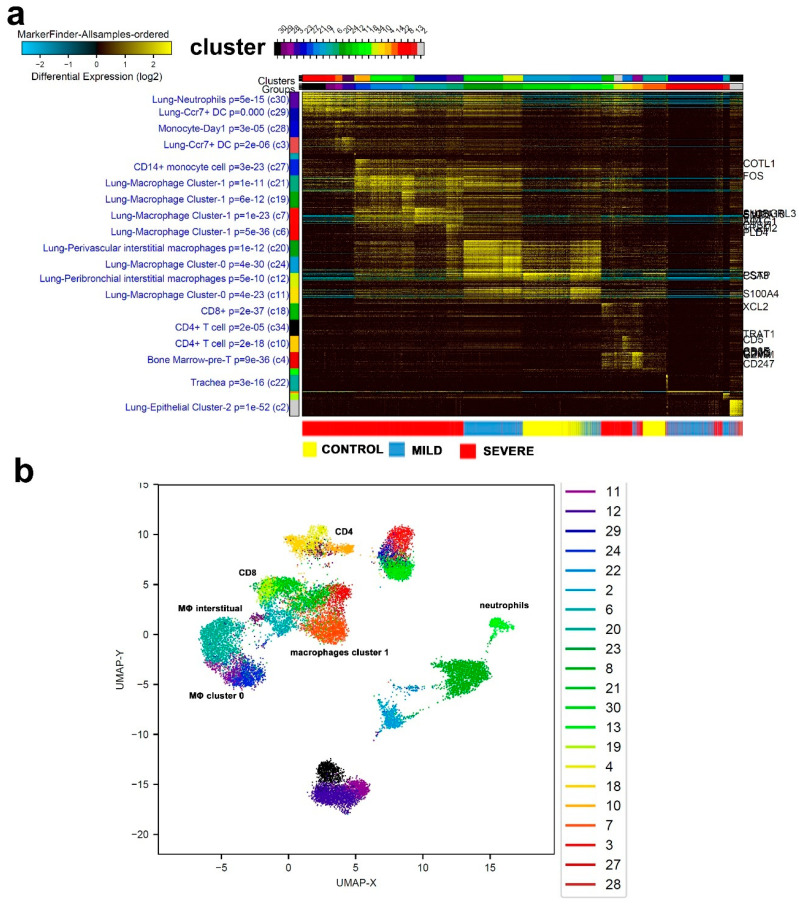
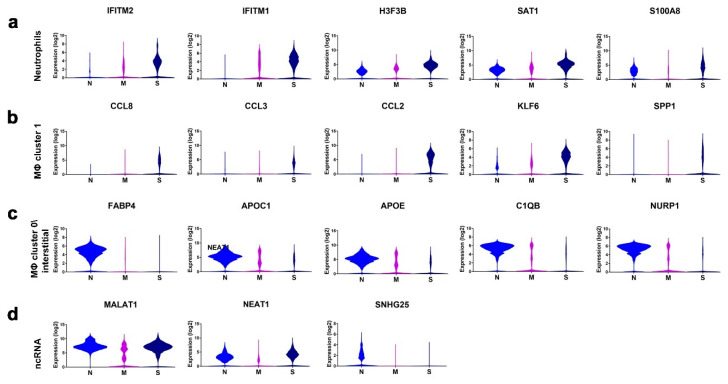
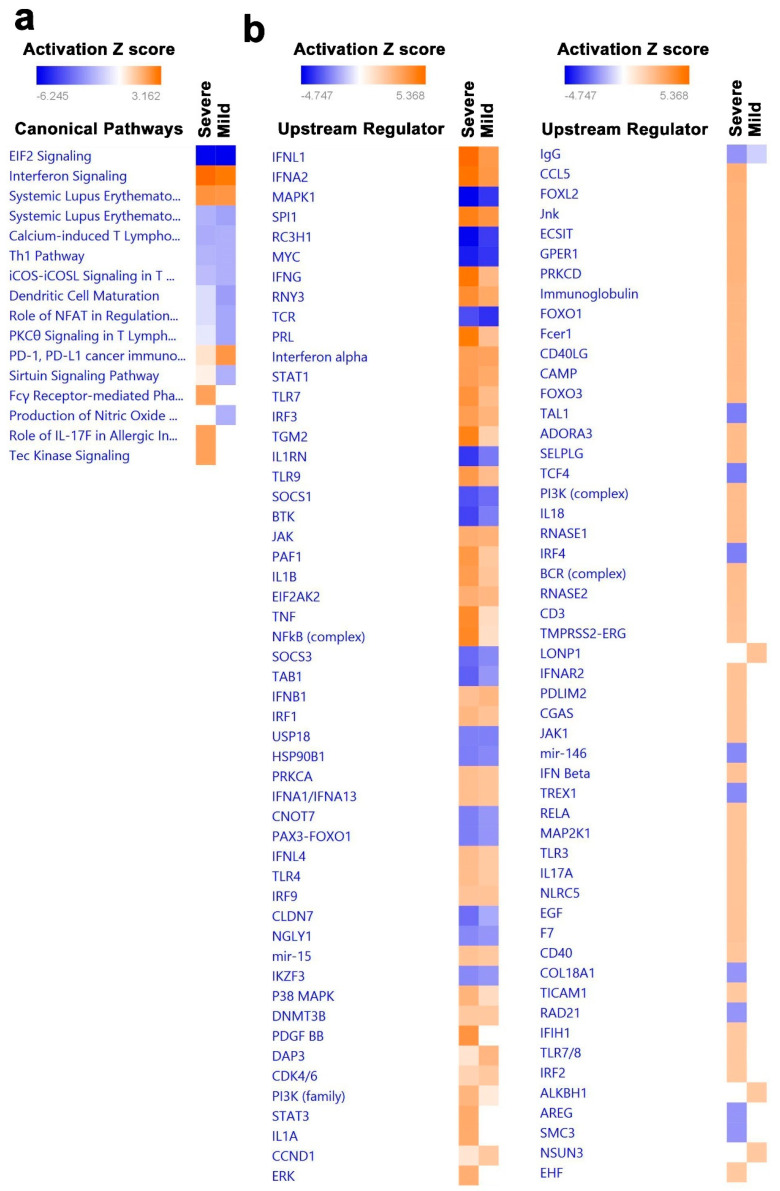
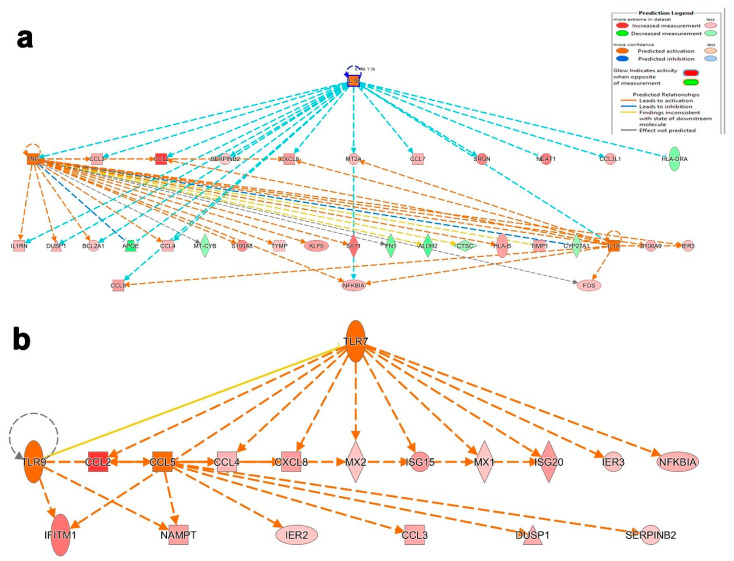
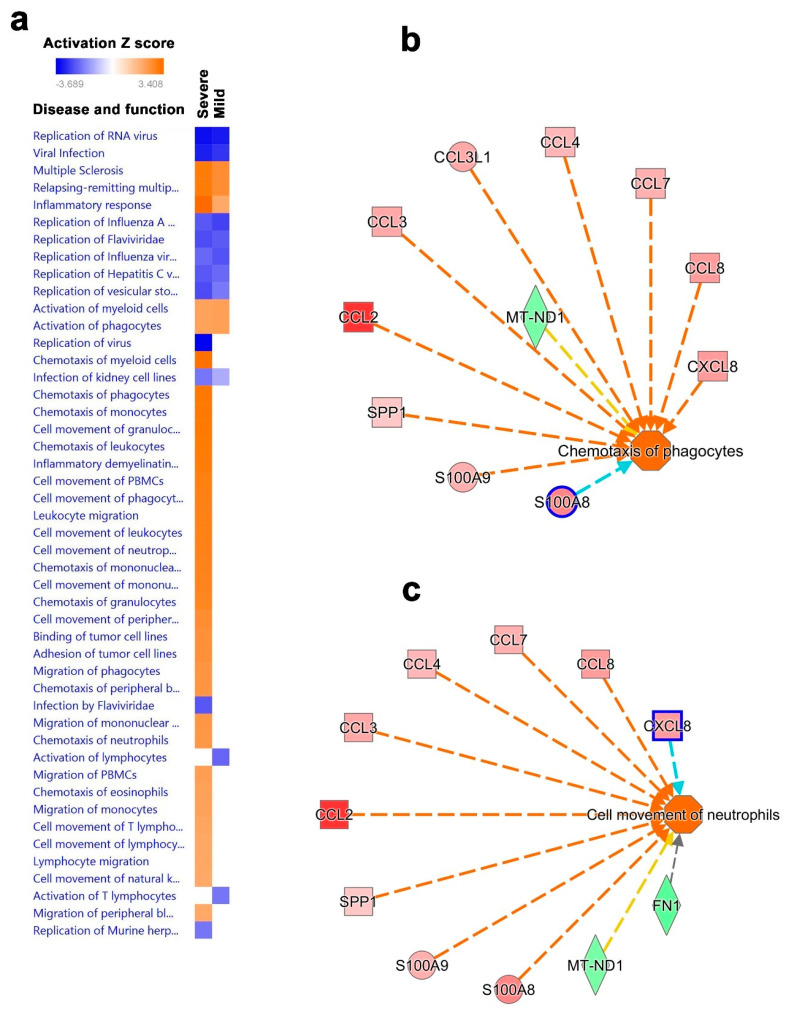
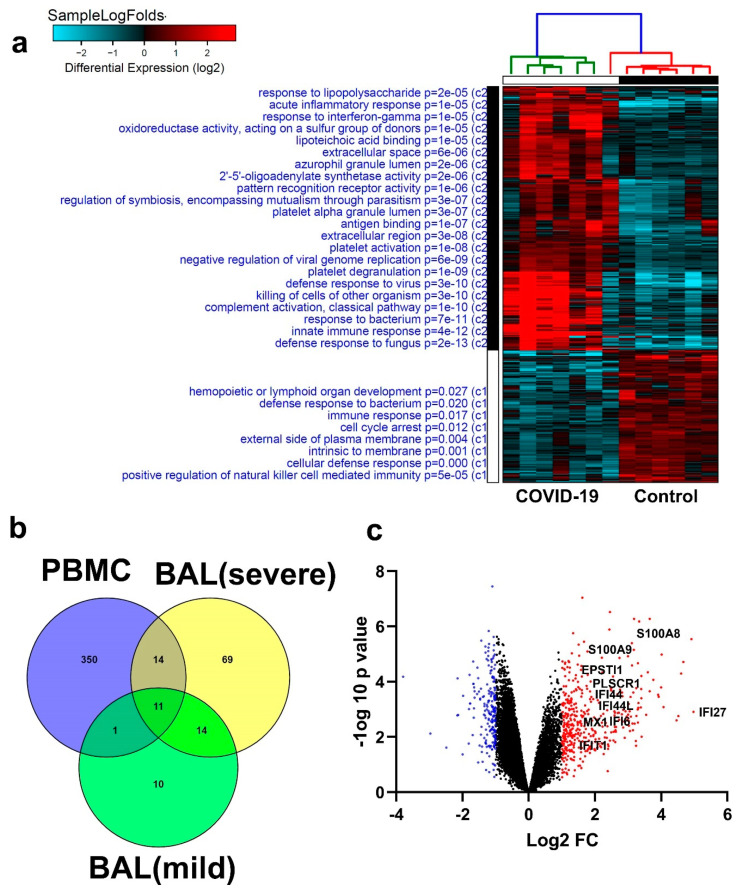
Cumulative data link cytokine storms with coronavirus disease 2019 (COVID-19) severity. The precise identification of immune cell subsets in bronchoalveolar lavage (BAL) and their correlation with COVID-19 disease severity are currently being unraveled. Herein, we employed iterative clustering and guide-gene selection 2 (ICGS2) as well as uniform manifold approximation and projection (UMAP) dimensionality reduction computational algorithms to decipher the complex immune and cellular composition of BAL, using publicly available datasets from a total of 68,873 single cells derived from two healthy subjects, three patients with mild COVID-19, and five patients with severe COVID-19. Our analysis revealed the presence of neutrophils and macrophage cluster-1 as a hallmark of severe COVID-19. Among the identified gene signatures, IFITM2, IFITM1, H3F3B, SAT1, and S100A8 gene signatures were highly associated with neutrophils, while CCL8, CCL3, CCL2, KLF6, and SPP1 were associated with macrophage cluster-1 in severe-COVID-19 patients. Interestingly, although macrophages were also present in healthy subjects and patients with mild COVID-19, they had different gene signatures, indicative of interstitial and cluster-0 macrophage (i.e., FABP4, APOC1, APOE, C1QB, and NURP1). Additionally, MALAT1, NEAT1, and SNGH25 were downregulated in patients with mild and severe COVID-19. Interferon signaling, FCγ receptor-mediated phagocytosis, IL17, and Tec kinase canonical pathways were enriched in patients with severe COVID-19, while PD-1 and PDL-1 pathways were suppressed. A number of upstream regulators (IFNG, PRL, TLR7, PRL, TGM2, TLR9, IL1B, TNF, NFkB, IL1A, STAT3, CCL5, and others) were also enriched in BAL cells from severe COVID-19-affected patients compared to those from patients with mild COVID-19. Further analyses revealed genes associated with the inflammatory response and chemotaxis of myeloid cells, phagocytes, and granulocytes, among the top activated functional categories in BAL from severe COVID-19-affected patients. Transcriptome data from another cohort of COVID-19-derived peripheral blood mononuclear cells (PBMCs) revealed the presence of several genes common to those found in BAL from patients with severe and mild COVID-19 (IFI27, IFITM3, IFI6, IFIT3, MX1, IFIT1, OASL, IFI30, OAS1) or to those seen only in BAL from severe-COVID-19 patients (S100A8, IFI44, IFI44L, CXCL8, CCR1, PLSCR1, EPSTI1, FPR1, OAS2, OAS3, IL1RN, TYMP, BCL2A1). Taken together, our data reveal the presence of neutrophils and macrophage cluster-1 as the main immune cell subsets associated with severe COVID-19 and identify their inflammatory and chemotactic gene signatures, also partially reflected systemically in the circulation, for possible diagnostic and therapeutic interventions.
Keywords: COVID-19; ICGS2; SARS-Cov-2; UMAP; bronchoalveolar lavage (BAL); inflammatory macrophages; neutrophils.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181 doi: 10.1016/j.cell.2020.02.052. - DOI - PMC - PubMed
-
- Jia H.P., Look D.C., Shi L., Hickey M., Pewe L., Netland J., Farzan M., Wohlford-Lenane C., Perlman S., McCray P.B., Jr. ACE2 Receptor Expression and Severe Acute Respiratory Syndrome Coronavirus Infection Depend on Differentiation of Human Airway Epithelia. J. Virol. 2005;79:14614–14621. doi: 10.1128/JVI.79.23.14614-14621.2005. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
