

Comparative roles of charge, π, and hydrophobic interactions in sequence-dependent phase separation of intrinsically disordered proteins
- PMID: 33139563
- PMCID: PMC7682375
- DOI: 10.1073/pnas.2008122117
Comparative roles of charge, π, and hydrophobic interactions in sequence-dependent phase separation of intrinsically disordered proteins
Abstract
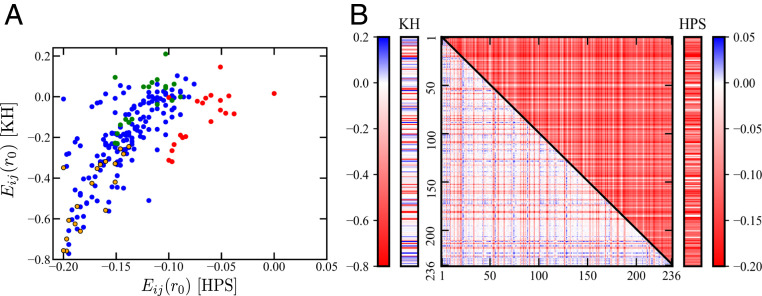
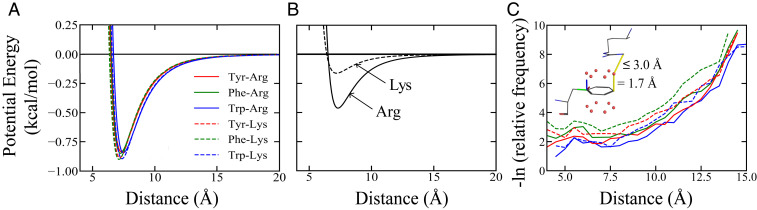
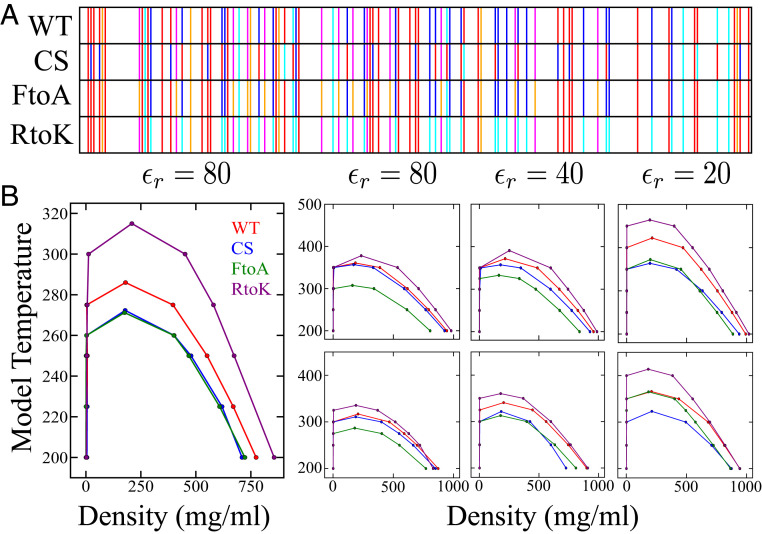
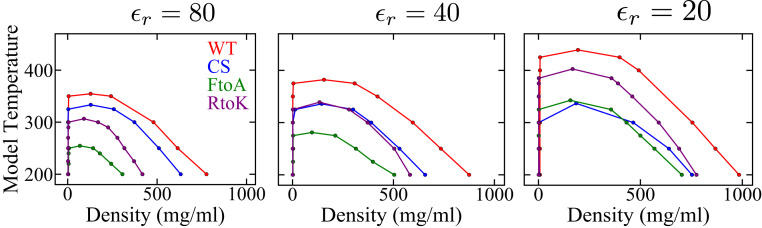
Endeavoring toward a transferable, predictive coarse-grained explicit-chain model for biomolecular condensates underlain by liquid-liquid phase separation (LLPS) of proteins, we conducted multiple-chain simulations of the N-terminal intrinsically disordered region (IDR) of DEAD-box helicase Ddx4, as a test case, to assess roles of electrostatic, hydrophobic, cation-π, and aromatic interactions in amino acid sequence-dependent LLPS. We evaluated three different residue-residue interaction schemes with a shared electrostatic potential. Neither a common hydrophobicity scheme nor one augmented with arginine/lysine-aromatic cation-π interactions consistently accounted for available experimental LLPS data on the wild-type, a charge-scrambled, a phenylalanine-to-alanine (FtoA), and an arginine-to-lysine (RtoK) mutant of Ddx4 IDR. In contrast, interactions based on contact statistics among folded globular protein structures reproduce the overall experimental trend, including that the RtoK mutant has a much diminished LLPS propensity. Consistency between simulation and experiment was also found for RtoK mutants of P-granule protein LAF-1, underscoring that, to a degree, important LLPS-driving π-related interactions are embodied in classical statistical potentials. Further elucidation is necessary, however, especially of phenylalanine's role in condensate assembly because experiments on FtoA and tyrosine-to-phenylalanine mutants suggest that LLPS-driving phenylalanine interactions are significantly weaker than posited by common statistical potentials. Protein-protein electrostatic interactions are modulated by relative permittivity, which in general depends on aqueous protein concentration. Analytical theory suggests that this dependence entails enhanced interprotein interactions in the condensed phase but more favorable protein-solvent interactions in the dilute phase. The opposing trends lead to only a modest overall impact on LLPS.
Keywords: biomolecular condensates; membraneless organelles; phase separation.
Conflict of interest statement
The authors declare no competing interest.
Figures
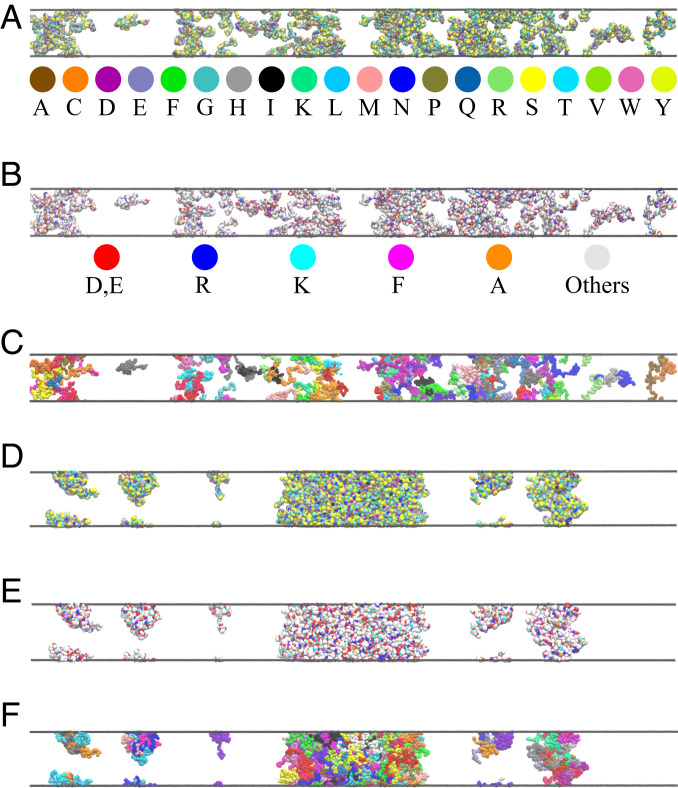
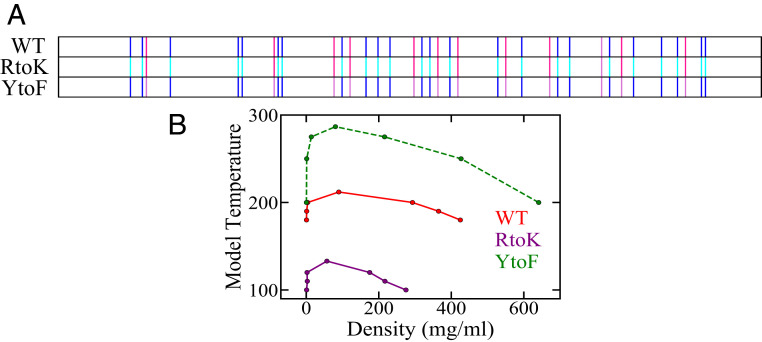
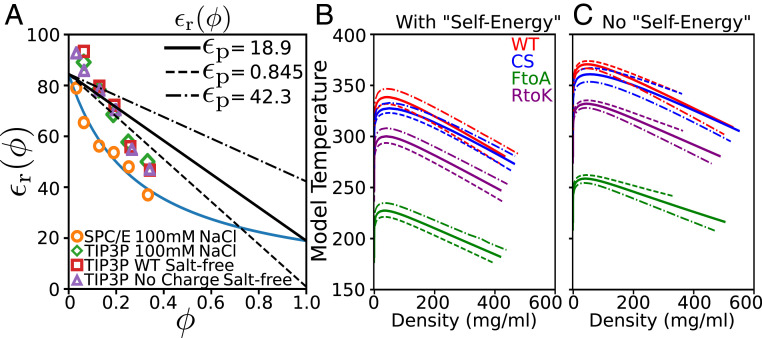
References
-
- Brangwynne C. P., et al. , Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
