

Construction of a complete set of Neisseria meningitidis mutants and its use for the phenotypic profiling of this human pathogen
- PMID: 33139723
- PMCID: PMC7606547
- DOI: 10.1038/s41467-020-19347-y
Construction of a complete set of Neisseria meningitidis mutants and its use for the phenotypic profiling of this human pathogen
Abstract
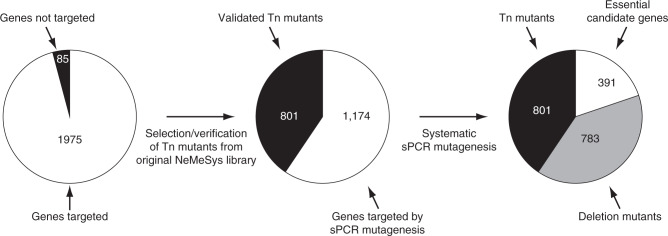
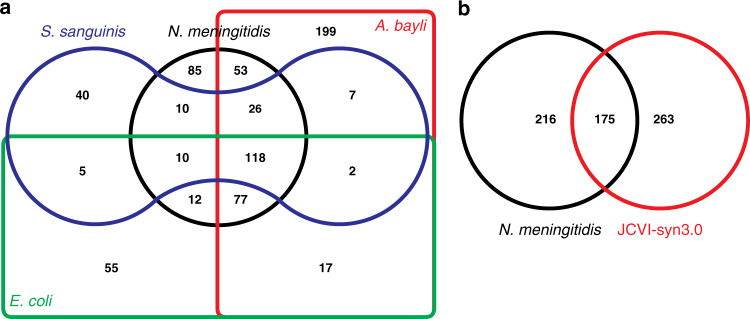
The bacterium Neisseria meningitidis causes life-threatening meningitis and sepsis. Here, we construct a complete collection of defined mutants in protein-coding genes of this organism, identifying all genes that are essential under laboratory conditions. The collection, named NeMeSys 2.0, consists of individual mutants in 1584 non-essential genes. We identify 391 essential genes, which are associated with basic functions such as expression and preservation of genome information, cell membrane structure and function, and metabolism. We use this collection to shed light on the functions of diverse genes, including a gene encoding a member of a previously unrecognised class of histidinol-phosphatases; a set of 20 genes required for type IV pili function; and several conditionally essential genes encoding antitoxins and/or immunity proteins. We expect that NeMeSys 2.0 will facilitate the phenotypic profiling of a major human bacterial pathogen.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
