

Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19
- PMID: 33142989
- PMCID: PMC7693583
- DOI: 10.3390/biomedicines8110460
Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19
Abstract
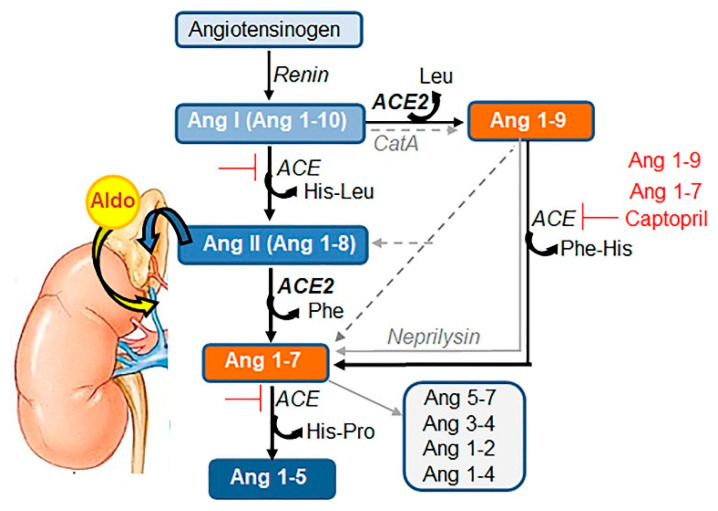
COVID-19 symptoms, including hypokalemia, hypoalbuminemia, ageusia, neurological dysfunctions, D-dimer production, and multi-organ microthrombosis reach beyond effects attributed to impaired angiotensin-converting enzyme 2 (ACE2) signaling and elevated concentrations of angiotensin II (Ang II). Although both SARS-CoV (Severe Acute Respiratory Syndrome Coronavirus) and SARS-CoV-2 utilize ACE2 for host entry, distinct COVID-19 pathogenesis coincides with the acquisition of a new sequence, which is homologous to the furin cleavage site of the human epithelial Na+ channel (ENaC). This review provides a comprehensive summary of the role of ACE2 in the assembly of Na+-dependent transporters of glucose, imino and neutral amino acids, as well as the functions of ENaC. Data support an osmotic adaptation mechanism in which osmotic and hemostatic instability induced by Ang II-activated ENaC is counterbalanced by an influx of organic osmolytes and Na+ through the ACE2 complex. We propose a paradigm for the two-site attack of SARS-CoV-2 leading to ENaC hyperactivation and inactivation of the ACE2 complex, which collapses cell osmolality and leads to rupture and/or necrotic death of swollen pulmonary, endothelial, and cardiac cells, thrombosis in infected and non-infected tissues, and aberrant sensory and neurological perception in COVID-19 patients. This dual mechanism employed by SARS-CoV-2 calls for combinatorial treatment strategies to address and prevent severe complications of COVID-19.
Keywords: angiotensin; coagulation; hypertension; inflammation; organ failure; thrombosis; tonicity; transporters; virus.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
