

The genetic landscape of axonal neuropathies in the middle-aged and elderly: Focus on MME
- PMID: 33144514
- PMCID: PMC7836667
- DOI: 10.1212/WNL.0000000000011132
The genetic landscape of axonal neuropathies in the middle-aged and elderly: Focus on MME
Abstract
Objective: To test the hypothesis that monogenic neuropathies such as Charcot-Marie-Tooth disease (CMT) contribute to frequent but often unexplained neuropathies in the elderly, we performed genetic analysis of 230 patients with unexplained axonal neuropathies and disease onset ≥35 years.
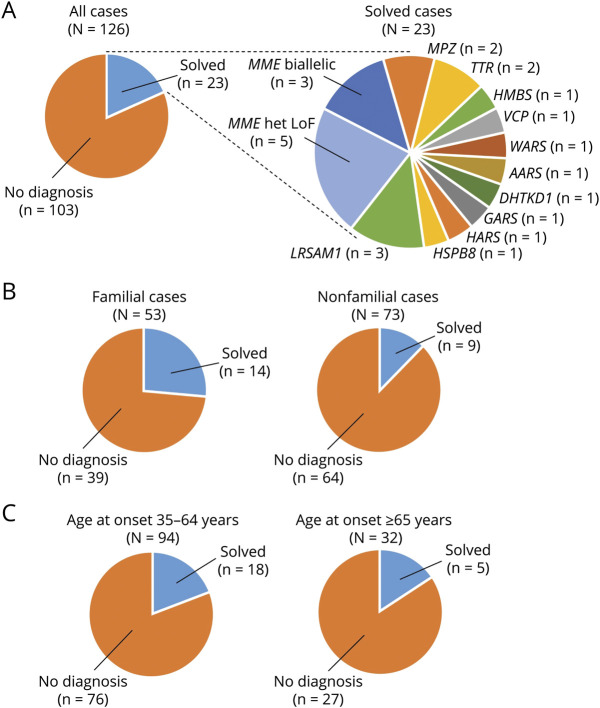
Methods: We recruited patients, collected clinical data, and conducted whole-exome sequencing (WES; n = 126) and MME single-gene sequencing (n = 104). We further queried WES repositories for MME variants and measured blood levels of the MME-encoded protein neprilysin.
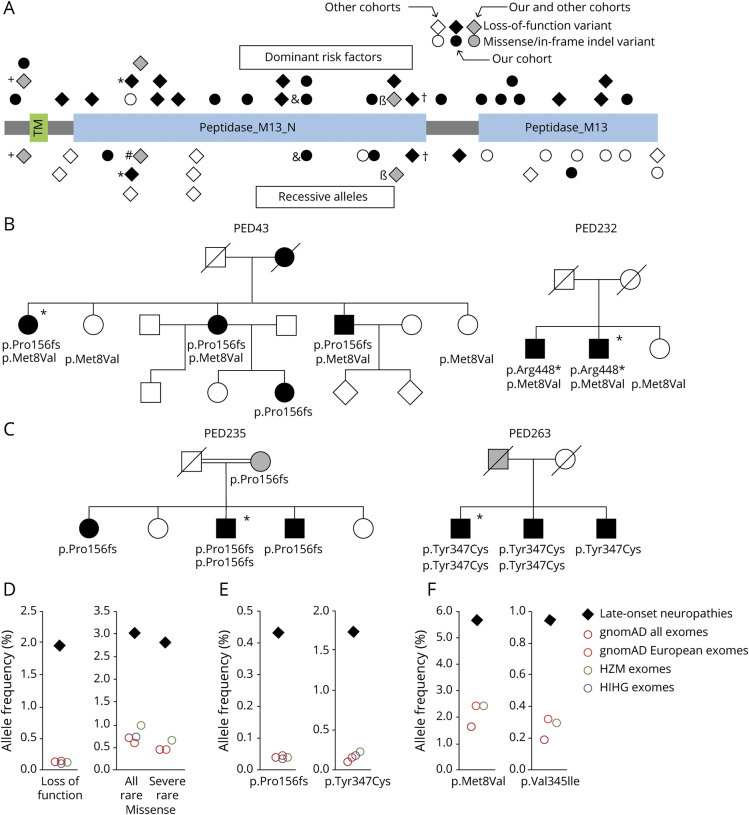
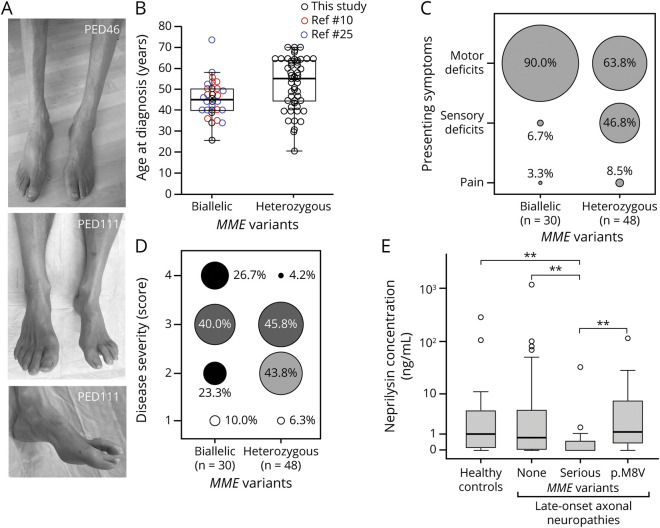
Results: In the WES cohort, the overall detection rate for assumed disease-causing variants in genes for CMT or other conditions associated with neuropathies was 18.3% (familial cases 26.4%, apparently sporadic cases 12.3%). MME was most frequently involved and accounted for 34.8% of genetically solved cases. The relevance of MME for late-onset neuropathies was further supported by detection of a comparable proportion of cases in an independent patient sample, preponderance of MME variants among patients compared to population frequencies, retrieval of additional late-onset neuropathy patients with MME variants from WES repositories, and low neprilysin levels in patients' blood samples. Transmission of MME variants was often consistent with an incompletely penetrant autosomal-dominant trait and less frequently with autosomal-recessive inheritance.
Conclusions: A detectable fraction of unexplained late-onset axonal neuropathies is genetically determined, by variants in either CMT genes or genes involved in other conditions that affect the peripheral nerves and can mimic a CMT phenotype. MME variants can act as completely penetrant recessive alleles but also confer dominantly inherited susceptibility to axonal neuropathies in an aging population.
© 2020 American Academy of Neurology.
Figures
Comment in
-
Genetics and adult-onset chronic idiopathic axonal neuropathy.Neurology. 2020 Dec 15;95(24):1071-1073. doi: 10.1212/WNL.0000000000011133. Epub 2020 Nov 3. Neurology. 2020. PMID: 33144513 No abstract available.
References
-
- Zis P, Sarrigiannis PG, Rao DG, Hewamadduma C, Hadjivassiliou M. Chronic idiopathic axonal polyneuropathy: a systematic review. J Neurol 2016;263:1903–1910. - PubMed
-
- Mathis S, Vallat JM, Ingrand P, Neau JP, Bouche P. Causes of neuropathy in the elderly: a retrospective study with 785 patients. Eur Geriatr Med 2015;6:114–118.
-
- Hanewinckel R, Drenthen J, van Oijen M, Hofman A, van Doorn PA, Ikram MA. Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016;87:1892–1898. - PubMed
-
- Auer-Grumbach M, Strasser-Fuchs S, Robl T, Windpassinger C, Wagner K. Late onset Charcot-Marie-Tooth 2 syndrome caused by two novel mutations in the MPZ gene. Neurology 2003;61:1435–1437. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous