

A trimeric Rab7 GEF controls NPC1-dependent lysosomal cholesterol export
- PMID: 33144569
- PMCID: PMC7642327
- DOI: 10.1038/s41467-020-19032-0
A trimeric Rab7 GEF controls NPC1-dependent lysosomal cholesterol export
Abstract
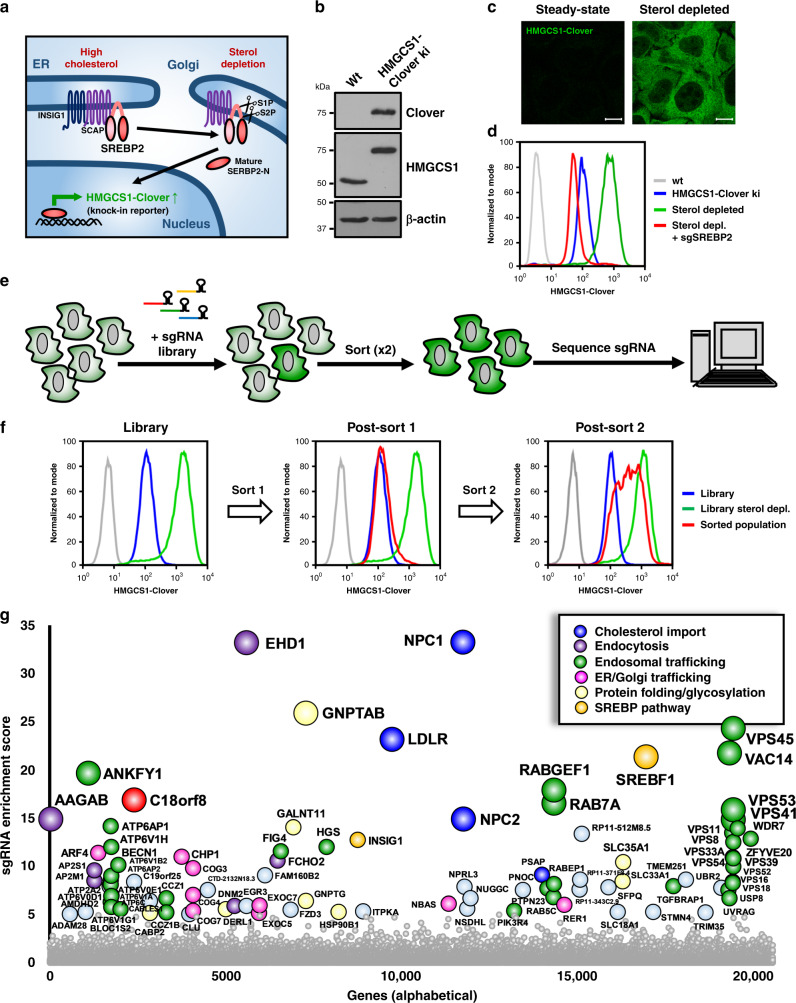
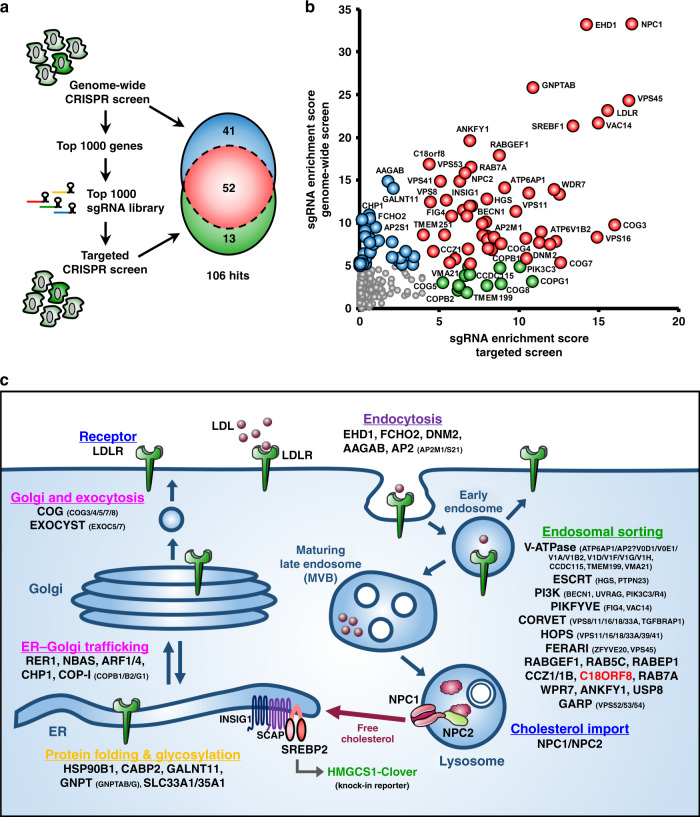
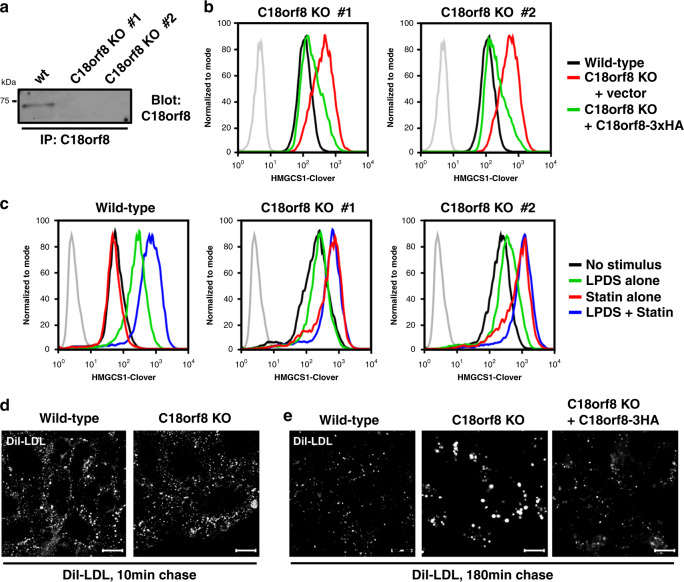
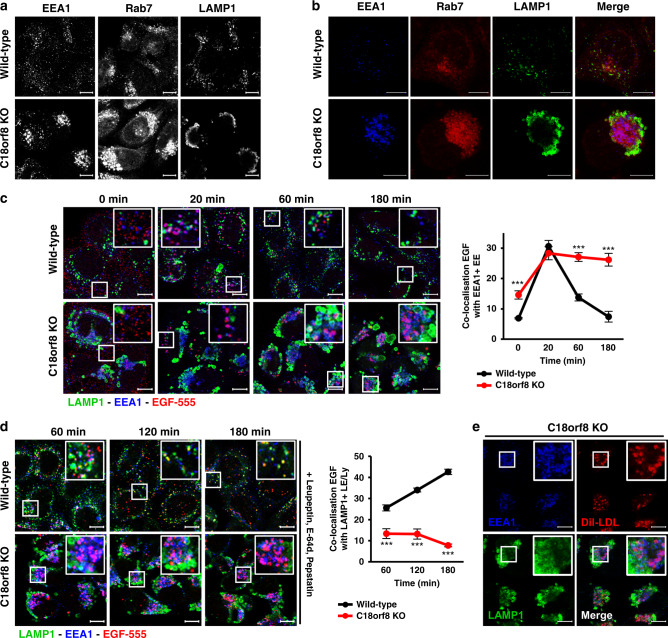
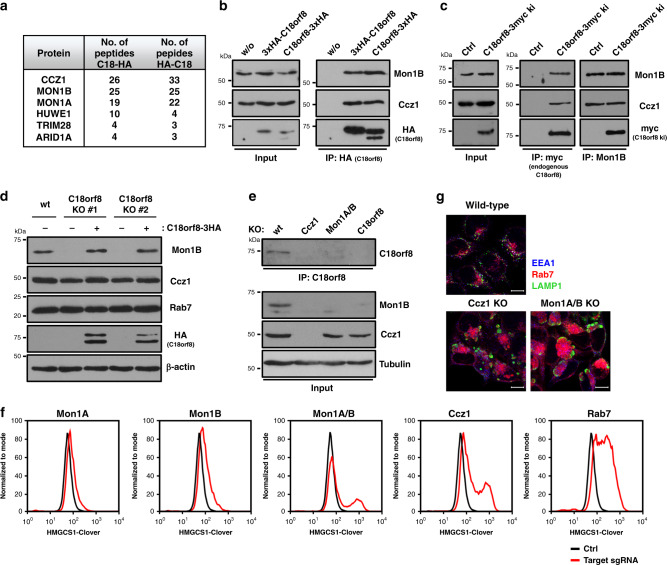
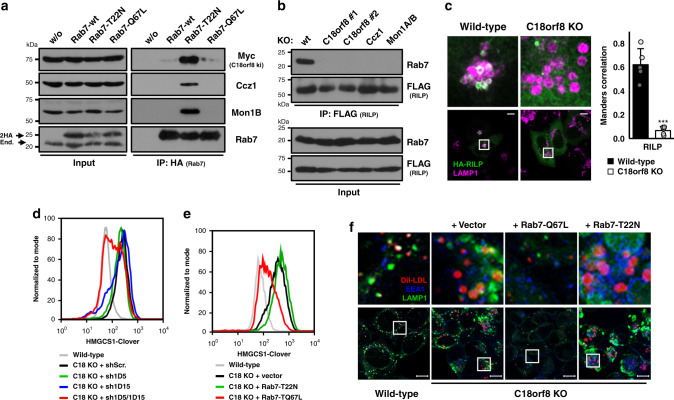
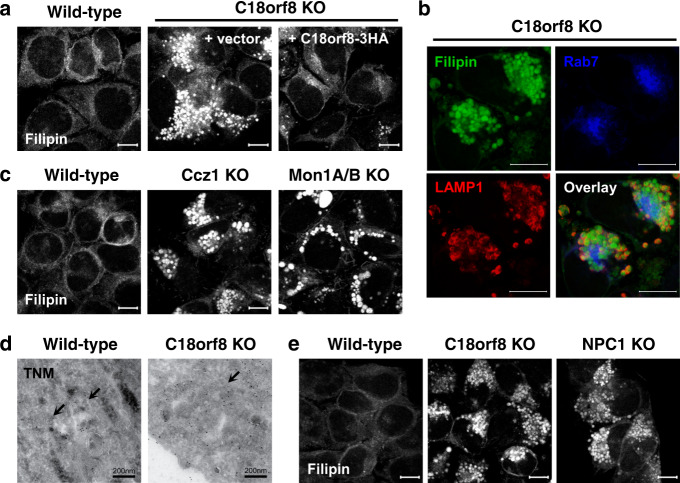
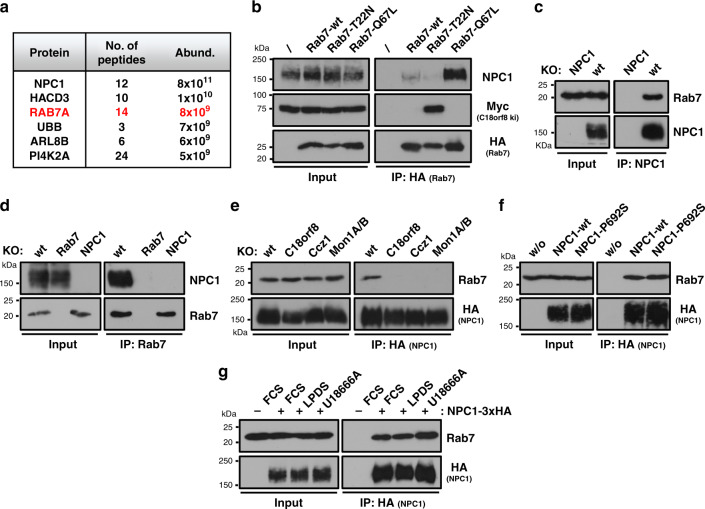
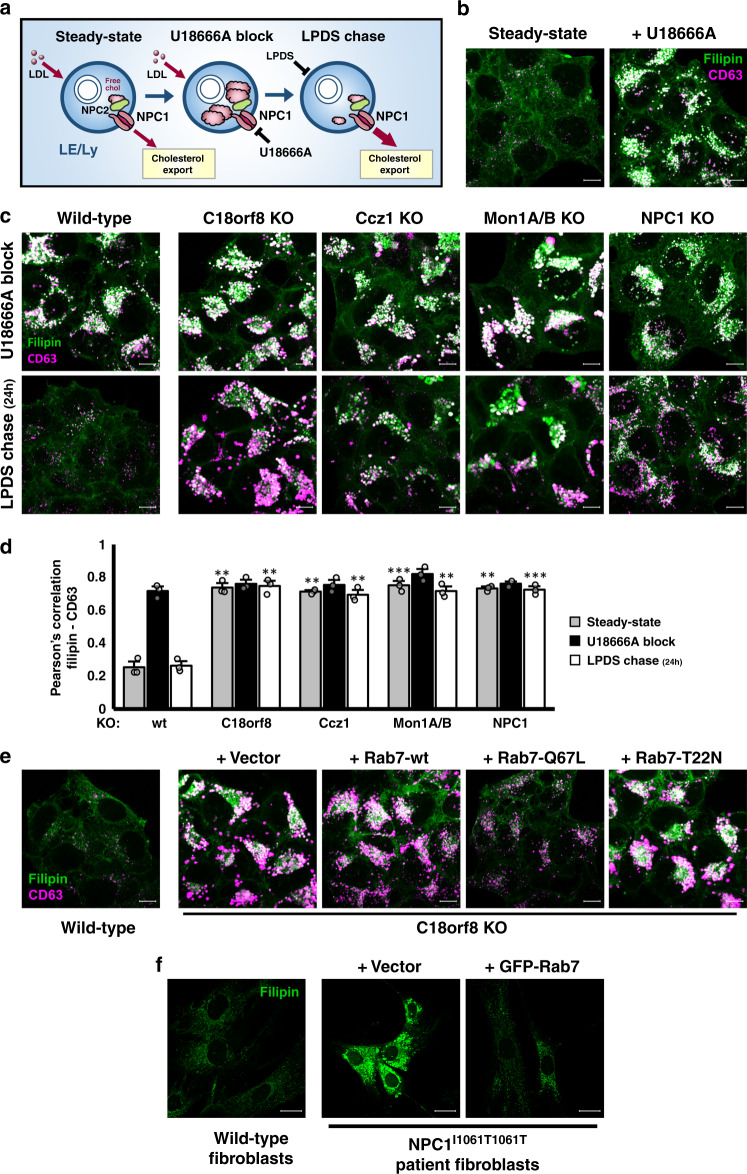
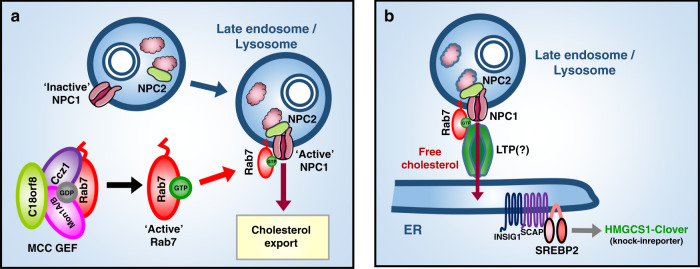
Cholesterol import in mammalian cells is mediated by the LDL receptor pathway. Here, we perform a genome-wide CRISPR screen using an endogenous cholesterol reporter and identify >100 genes involved in LDL-cholesterol import. We characterise C18orf8 as a core subunit of the mammalian Mon1-Ccz1 guanidine exchange factor (GEF) for Rab7, required for complex stability and function. C18orf8-deficient cells lack Rab7 activation and show severe defects in late endosome morphology and endosomal LDL trafficking, resulting in cellular cholesterol deficiency. Unexpectedly, free cholesterol accumulates within swollen lysosomes, suggesting a critical defect in lysosomal cholesterol export. We find that active Rab7 interacts with the NPC1 cholesterol transporter and licenses lysosomal cholesterol export. This process is abolished in C18orf8-, Ccz1- and Mon1A/B-deficient cells and restored by a constitutively active Rab7. The trimeric Mon1-Ccz1-C18orf8 (MCC) GEF therefore plays a central role in cellular cholesterol homeostasis coordinating Rab7 activation, endosomal LDL trafficking and NPC1-dependent lysosomal cholesterol export.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
