

Clinical Interpretation and Management of Genetic Variants
- PMID: 33145465
- PMCID: PMC7591931
- DOI: 10.1016/j.jacbts.2020.05.013
Clinical Interpretation and Management of Genetic Variants
Abstract
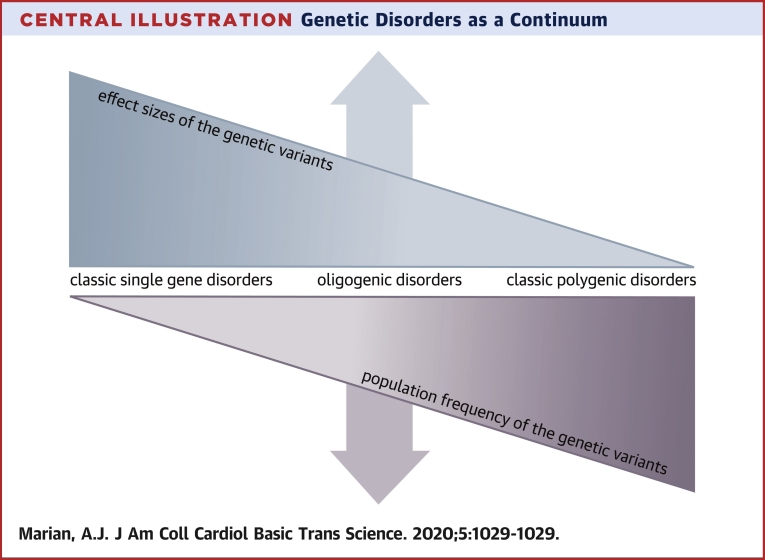
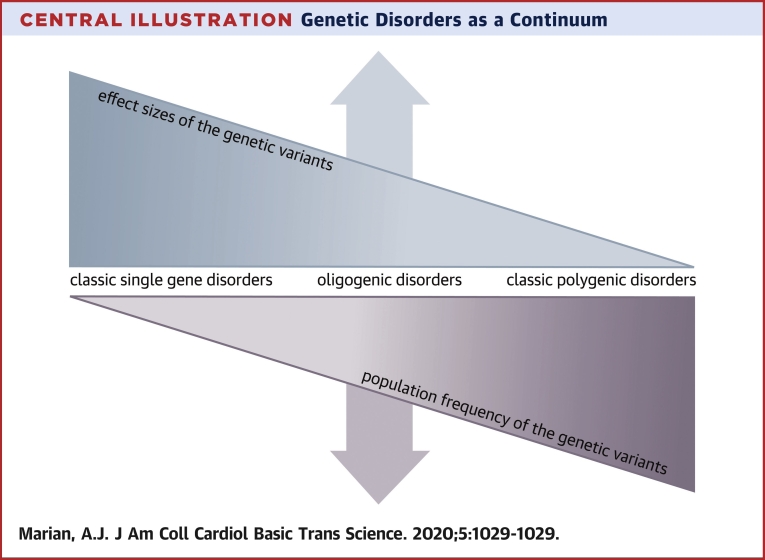
Genetic variants are major determinants of susceptibility to disease, response to therapy, and clinical outcomes. Advances in the short-read sequencing technologies, despite some shortcomings, have enabled identification of the vast majority of the genetic variants in each genome. The major challenge is in identifying the pathogenic variants in cardiovascular diseases. The yield of the genetic testing has been limited because of technological shortcomings and our incomplete understanding of the genetic basis of cardiovascular disorders. To advance the field, a shift to long-read sequencing platforms is necessary. In addition, to discern the pathogenic variants, genetic diseases should be considered as a continuum and the genetic variants as probabilistic factors with a gradient of effect sizes. Moreover, disease-specific physician-scientists with expertise in the clinical medicine and molecular genetics are best equipped to discern functional and clinical significance of the genetic variants. The changes would be expected to enhance clinical utilities of the genetic discoveries.
Keywords: CNV, copy number variants; HCM, hypertrophic cardiomyopathy; LoF, loss of function; SNV, single nucleotide variant; SV, structural variant; WES, whole exome sequencing; WGS, whole genome sequencing; genetic testing; genetic variants; indel, insertion/deletion; nsSNV, nonsynonymous single nucleotide variant; sequencing.
© 2020 The Author.
Conflict of interest statement
This work was supported in part by National Institutes of Health grant S10 OD018135, National Heart, Lung, and Blood Institute grants R01 HL151737, 1R01HL132401, and S10OD018135 Leducq Foundation grant 14 CVD 03, The Ewing Halsell Foundation, George and Mary Josephine Hamman Foundation, and the TexGen Fund from Greater Houston Community Foundation. Dr. Marian has reported that he has no relationships relevant to the contents of this paper to disclose.
Figures
References
-
- Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
