

Population genomics for wildlife conservation and management
- PMID: 33145846
- PMCID: PMC7894518
- DOI: 10.1111/mec.15720
Population genomics for wildlife conservation and management
Abstract
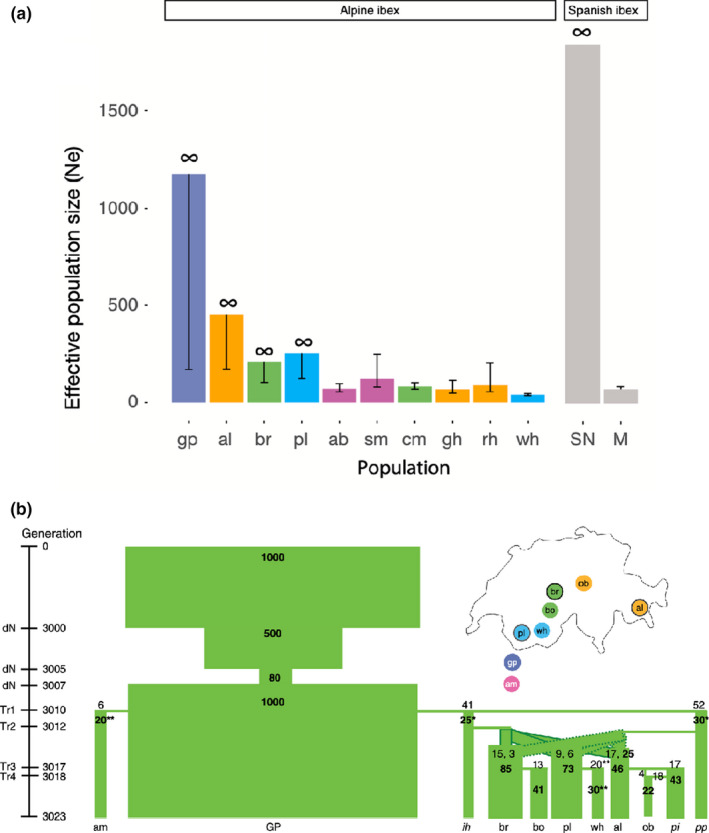
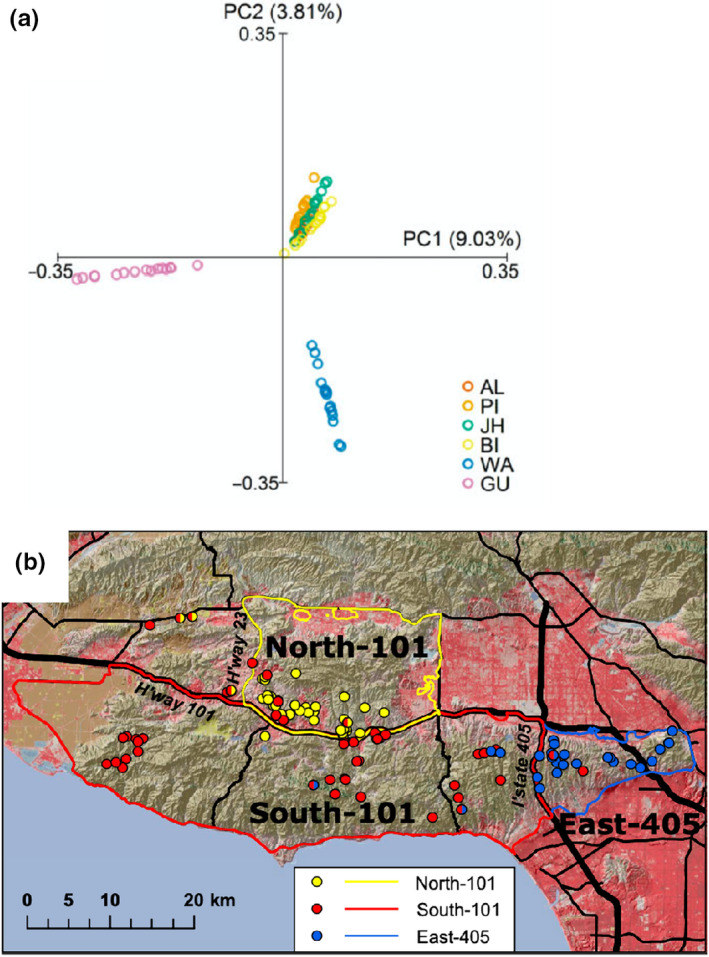
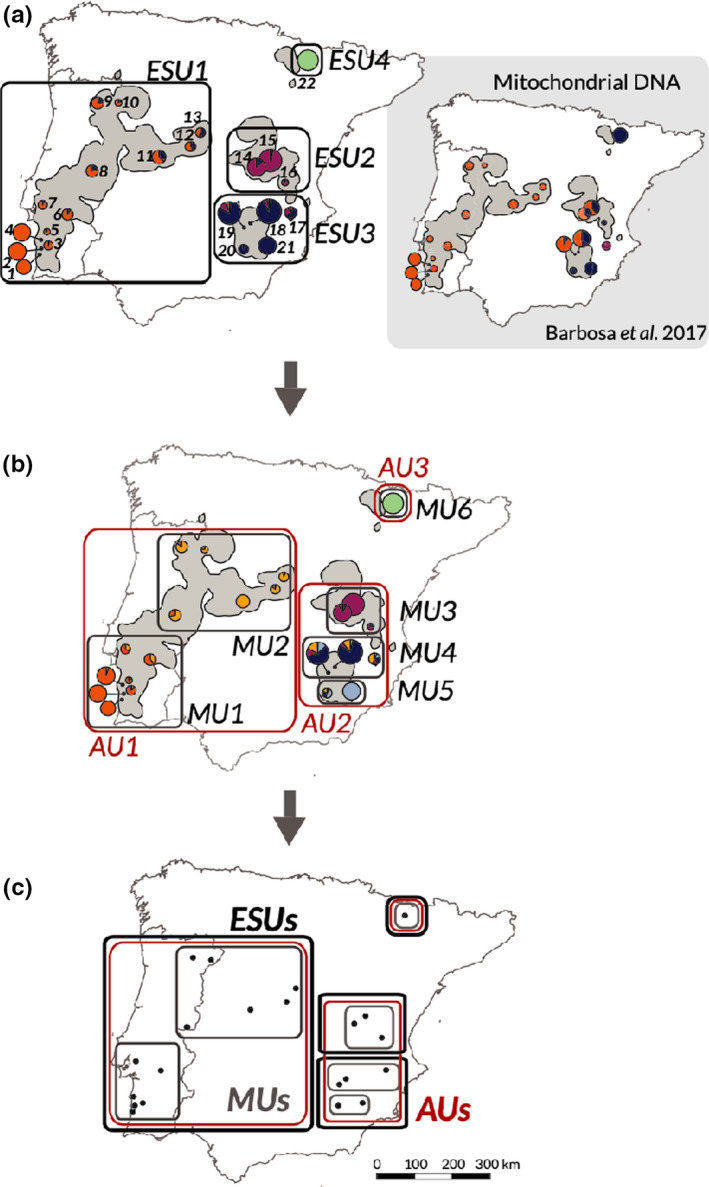
Biodiversity is under threat worldwide. Over the past decade, the field of population genomics has developed across nonmodel organisms, and the results of this research have begun to be applied in conservation and management of wildlife species. Genomics tools can provide precise estimates of basic features of wildlife populations, such as effective population size, inbreeding, demographic history and population structure, that are critical for conservation efforts. Moreover, population genomics studies can identify particular genetic loci and variants responsible for inbreeding depression or adaptation to changing environments, allowing for conservation efforts to estimate the capacity of populations to evolve and adapt in response to environmental change and to manage for adaptive variation. While connections from basic research to applied wildlife conservation have been slow to develop, these connections are increasingly strengthening. Here we review the primary areas in which population genomics approaches can be applied to wildlife conservation and management, highlight examples of how they have been used, and provide recommendations for building on the progress that has been made in this field.
Keywords: adaptive capacity; conservation units; effective population size; genetic rescue; inbreeding depression; population connectivity.
© 2020 The Authors. Molecular Ecology published by 2020 John Wiley & Sons Ltd.
Figures
References
-
- Allendorf, F. W. , Leary, R. F. , Spruell, P. , & Wenburg, J. K. (2001). The problems with hybrids: Setting conservation guidelines. Trends in Ecology and Evolution, 16, 613–622. 10.1016/S0169-5347(01)02290-X - DOI
-
- Andrews, K. R. , deBarba, M. , Russello, M. A. , & Waits, L. P. (2018). Advances in using non‐invasive, archival, and environmental samples for population genomic studies In Rajora O. P. (Ed.), Population Genomics: Wildlife. Springer Nature Switzerland AG.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
