

Computational strategies to combat COVID-19: useful tools to accelerate SARS-CoV-2 and coronavirus research
- PMID: 33147627
- PMCID: PMC7665365
- DOI: 10.1093/bib/bbaa232
Computational strategies to combat COVID-19: useful tools to accelerate SARS-CoV-2 and coronavirus research
Abstract
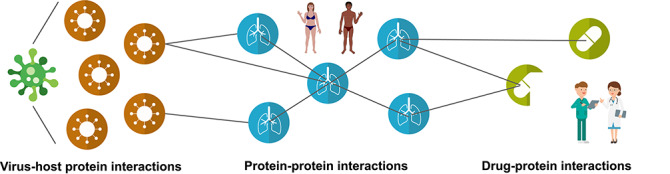
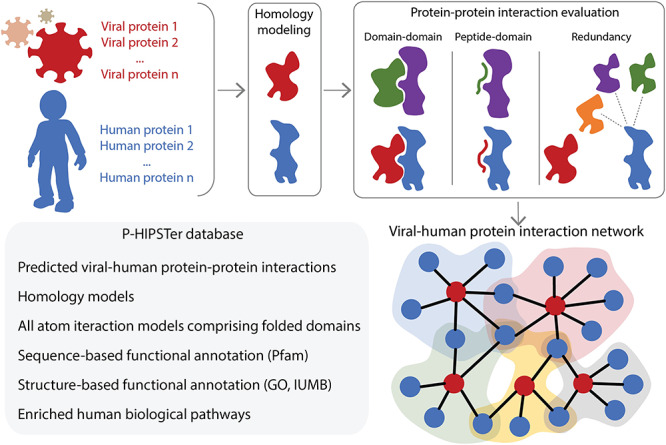
SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is a novel virus of the family Coronaviridae. The virus causes the infectious disease COVID-19. The biology of coronaviruses has been studied for many years. However, bioinformatics tools designed explicitly for SARS-CoV-2 have only recently been developed as a rapid reaction to the need for fast detection, understanding and treatment of COVID-19. To control the ongoing COVID-19 pandemic, it is of utmost importance to get insight into the evolution and pathogenesis of the virus. In this review, we cover bioinformatics workflows and tools for the routine detection of SARS-CoV-2 infection, the reliable analysis of sequencing data, the tracking of the COVID-19 pandemic and evaluation of containment measures, the study of coronavirus evolution, the discovery of potential drug targets and development of therapeutic strategies. For each tool, we briefly describe its use case and how it advances research specifically for SARS-CoV-2. All tools are free to use and available online, either through web applications or public code repositories. Contact:evbc@unj-jena.de.
Keywords: SARS-CoV-2; drug design; epidemiology; sequencing; tools; virus bioinformatics.
© The Author(s) 2020. Published by Oxford University Press.
Figures
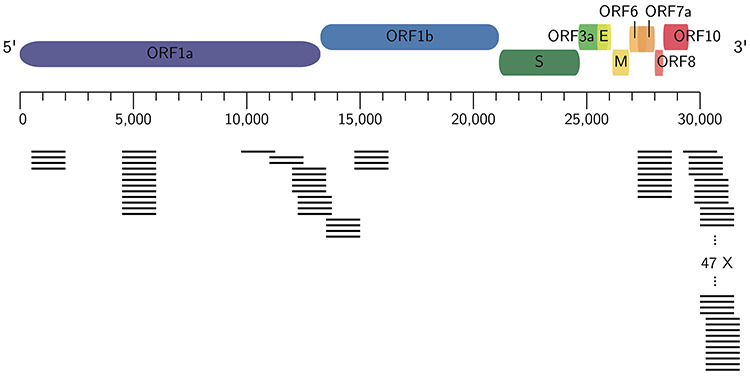
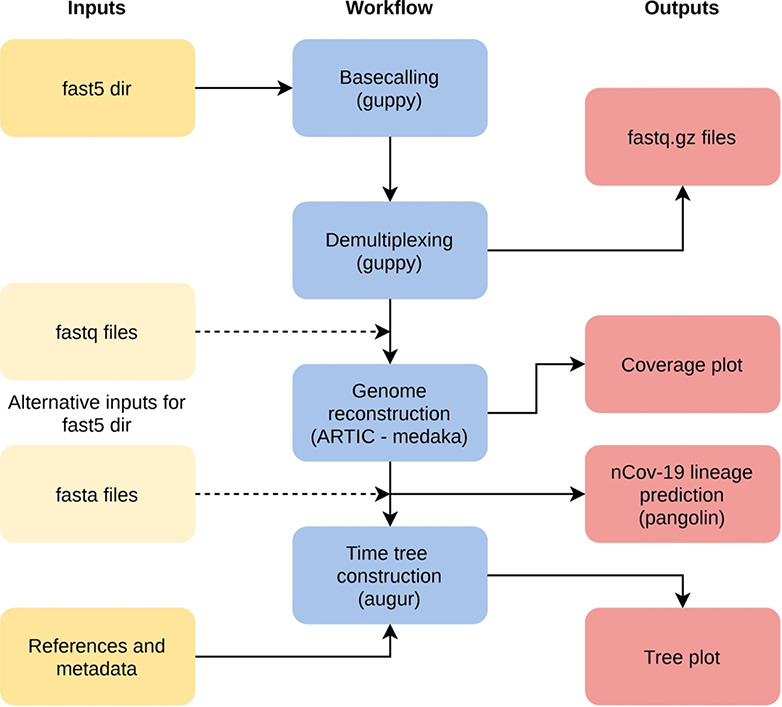
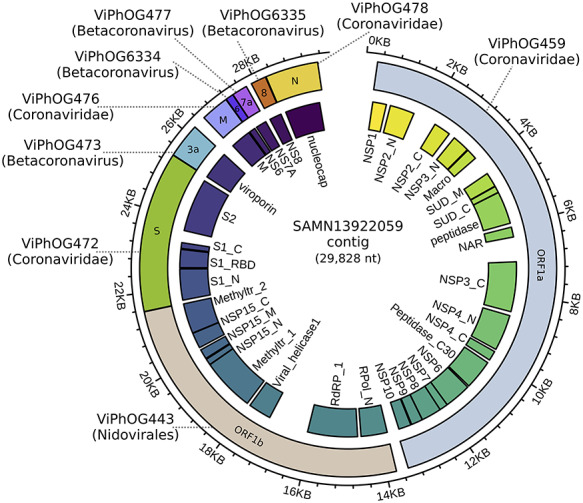
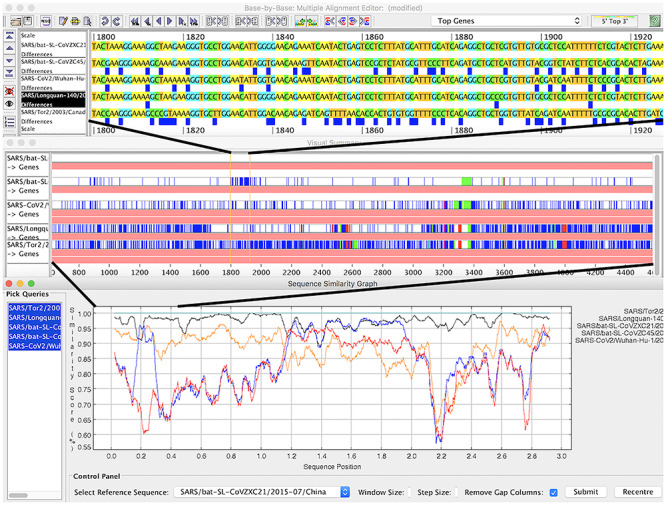
References
-
- Aguilera LU, Rodríguez-González J. Modeling the effect of tat inhibitors on HIV latency. J Theor Biol 2019;473:20–7. - PubMed
-
- Akgül A, Khoshnaw SHA, Mohammed WH. Mathematical model for the ebola virus disease. J Adv Phys 2018;7(2):190–8.
-
- Barry J. K.. Mathematical modelling of the HIV life cycle: identifying optimal treatment strategies. PhD thesis, University of Greifswald, 2018.
-
- Boettiger C.. An introduction to docker for reproducible research. ACM SIGOPS Operating Systems Review, 2015;49(1):71–79.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
