

iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy
- PMID: 33148626
- PMCID: PMC8861807
- DOI: 10.1126/scitranslmed.aaz5618
iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy
Abstract
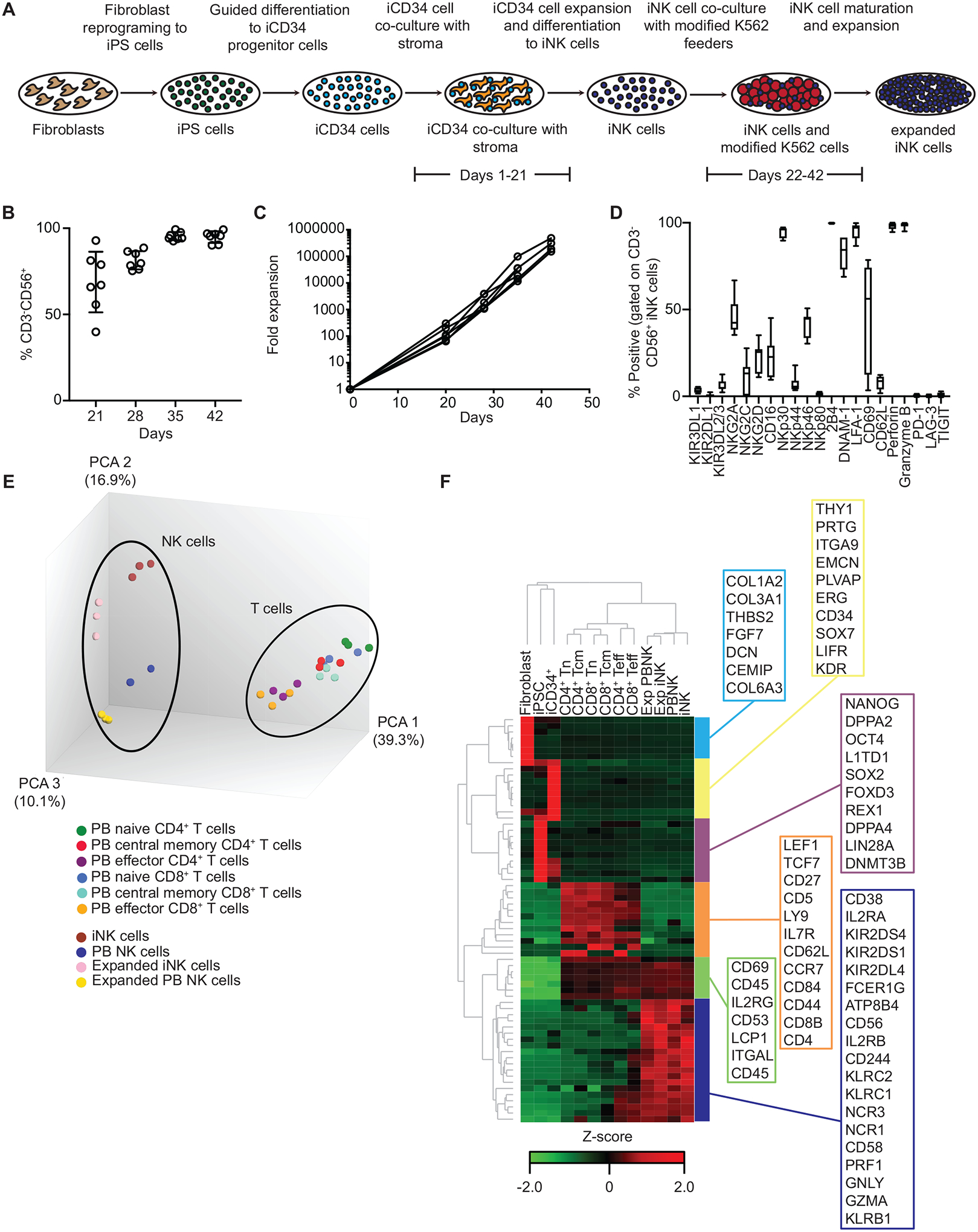
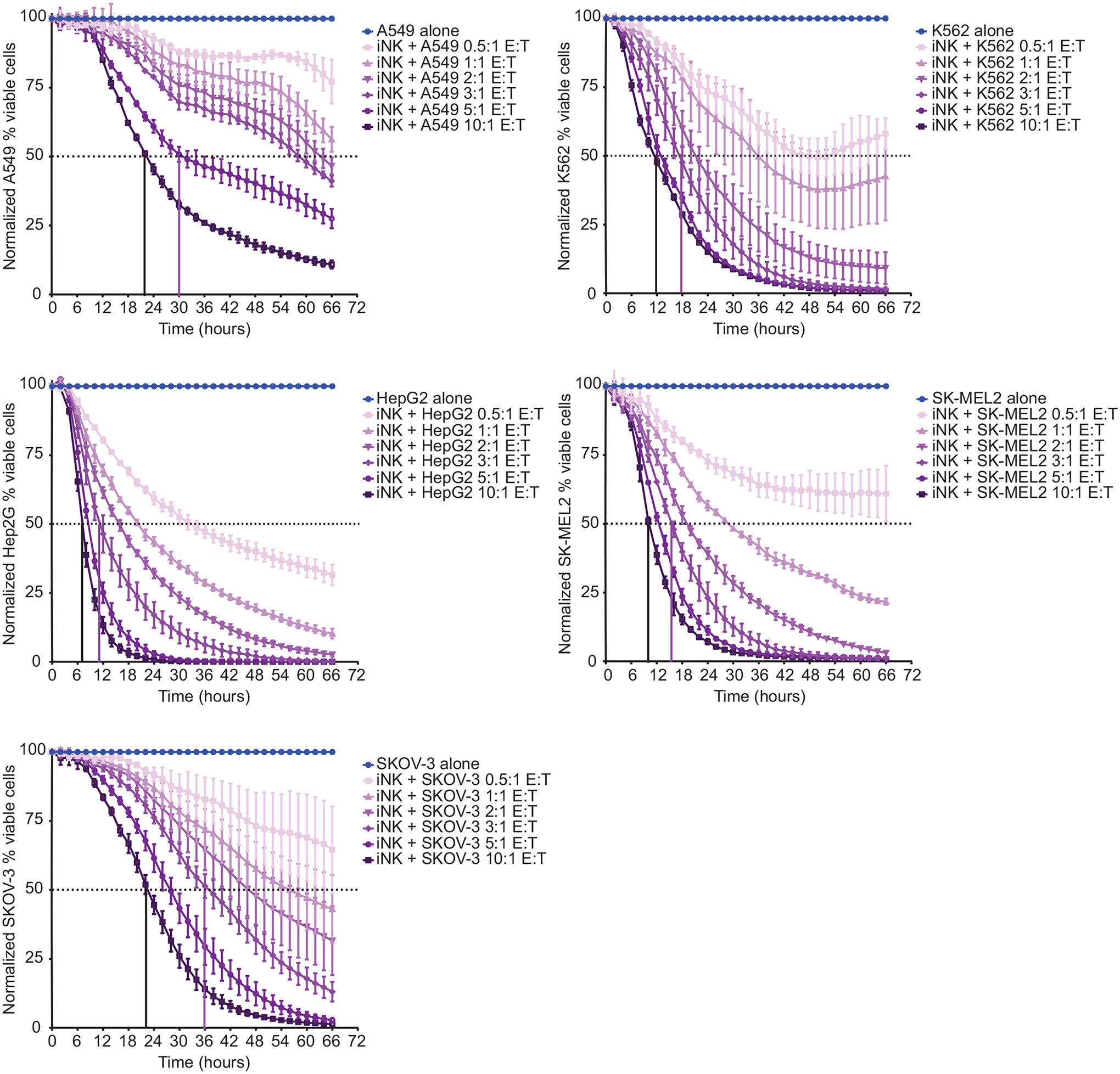
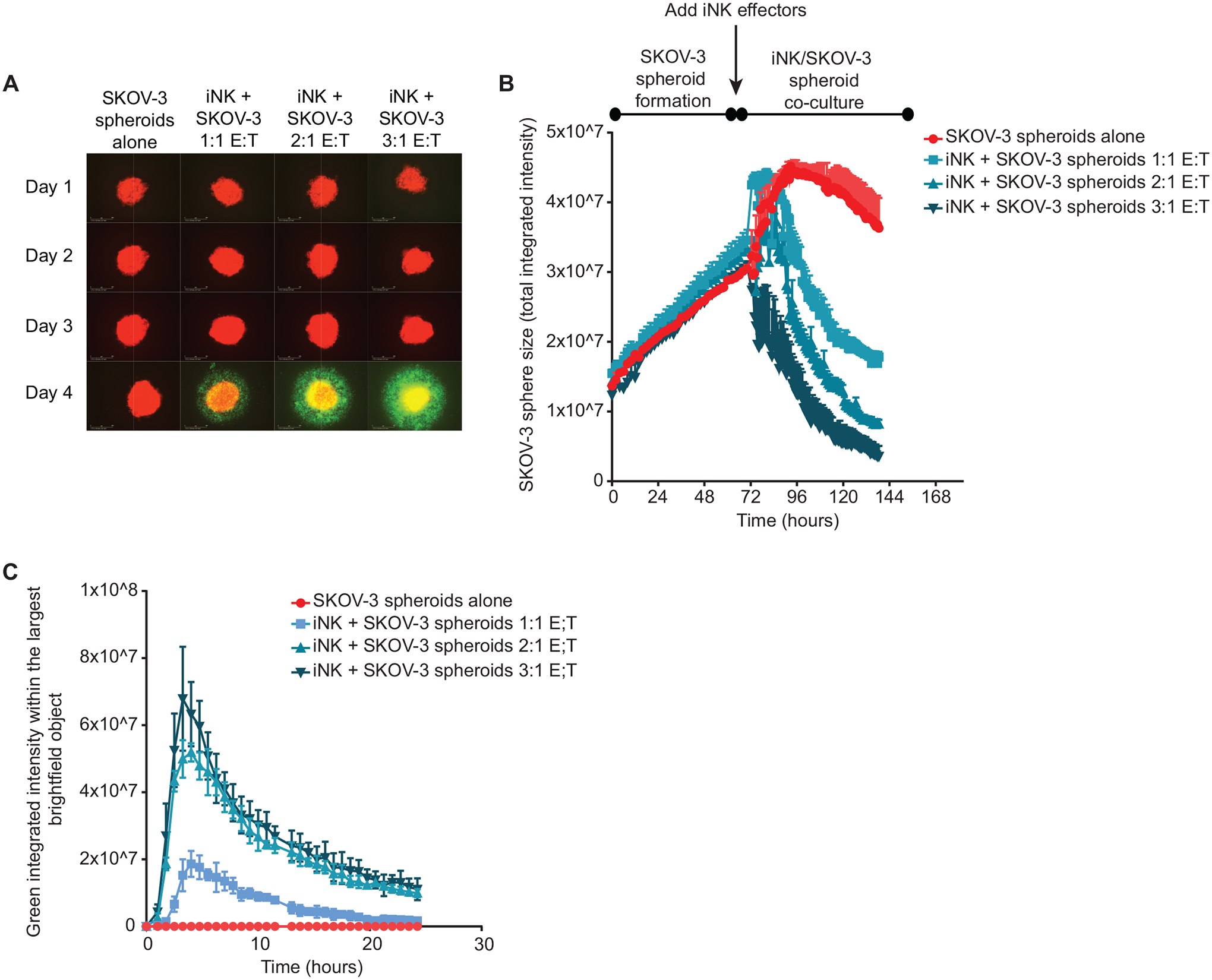
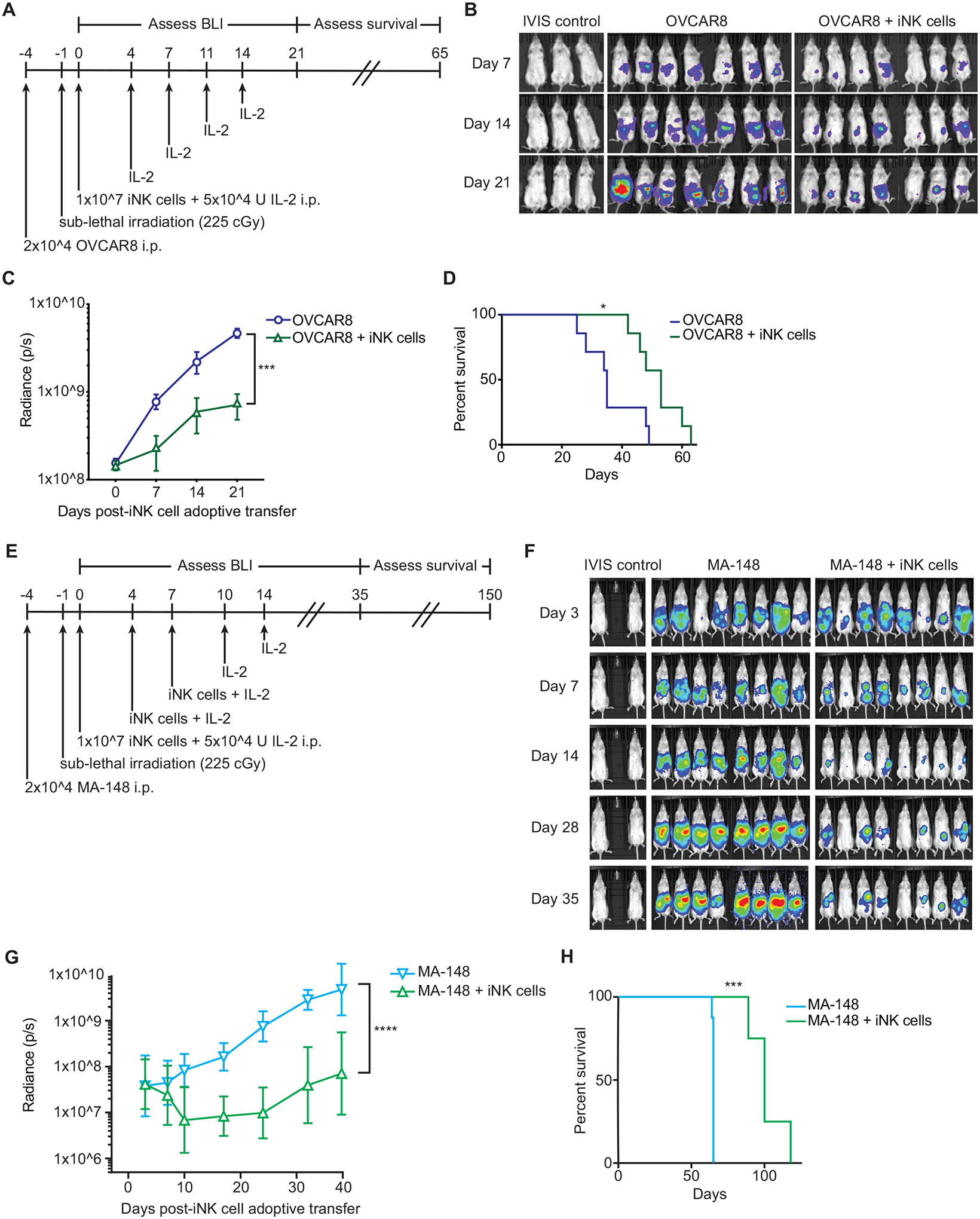
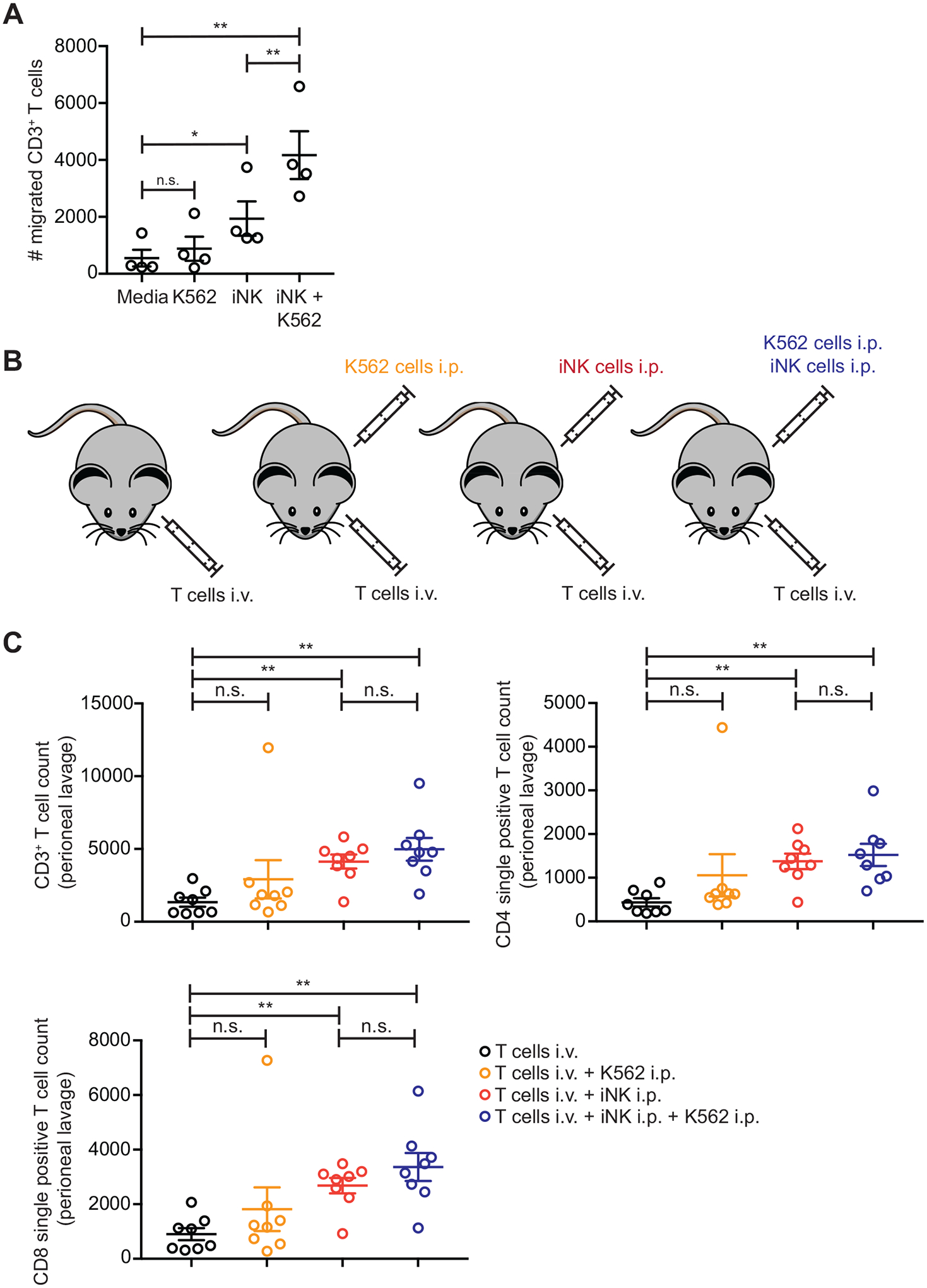
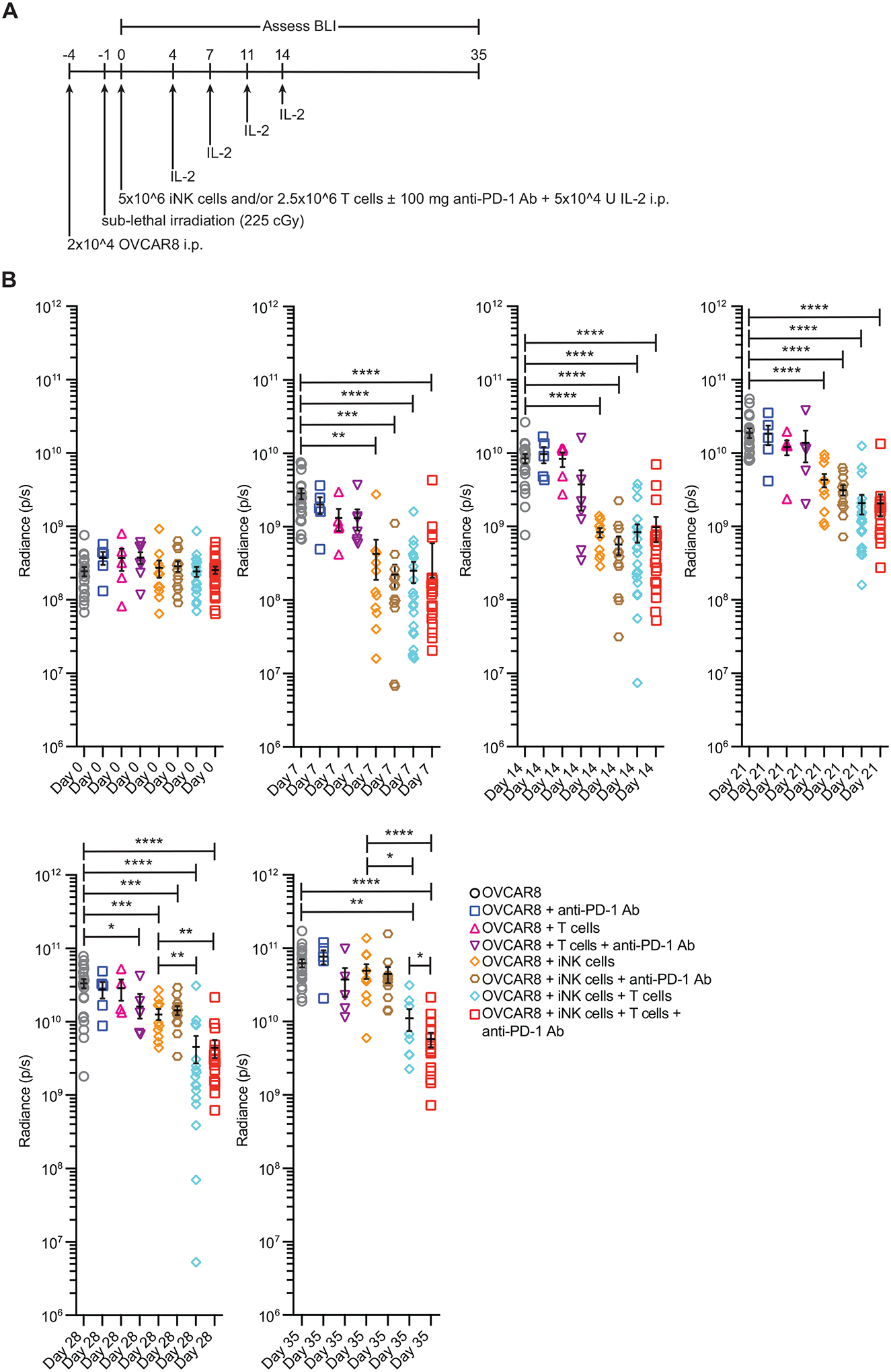
The development of immunotherapeutic monoclonal antibodies targeting checkpoint inhibitory receptors, such as programmed cell death 1 (PD-1), or their ligands, such as PD-L1, has transformed the oncology landscape. However, durable tumor regression is limited to a minority of patients. Therefore, combining immunotherapies with those targeting checkpoint inhibitory receptors is a promising strategy to bolster antitumor responses and improve response rates. Natural killer (NK) cells have the potential to augment checkpoint inhibition therapies, such as PD-L1/PD-1 blockade, because NK cells mediate both direct tumor lysis and T cell activation and recruitment. However, sourcing donor-derived NK cells for adoptive cell therapy has been limited by both cell number and quality. Thus, we developed a robust and efficient manufacturing system for the differentiation and expansion of high-quality NK cells derived from induced pluripotent stem cells (iPSCs). iPSC-derived NK (iNK) cells produced inflammatory cytokines and exerted strong cytotoxicity against an array of hematologic and solid tumors. Furthermore, we showed that iNK cells recruit T cells and cooperate with T cells and anti-PD-1 antibody, further enhancing inflammatory cytokine production and tumor lysis. Because the iNK cell derivation process uses a renewable starting material and enables the manufacturing of large numbers of doses from a single manufacture, iNK cells represent an "off-the-shelf" source of cells for immunotherapy with the capacity to target tumors and engage the adaptive arm of the immune system to make a "cold" tumor "hot" by promoting the influx of activated T cells to augment checkpoint inhibitor therapies.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures

References
-
- Wherry EJ, T cell exhaustion. Nat. Immunol 12, 492–499 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
