

Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons
- PMID: 33149216
- PMCID: PMC7643171
- DOI: 10.1038/s41467-020-19319-2
Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons
Abstract
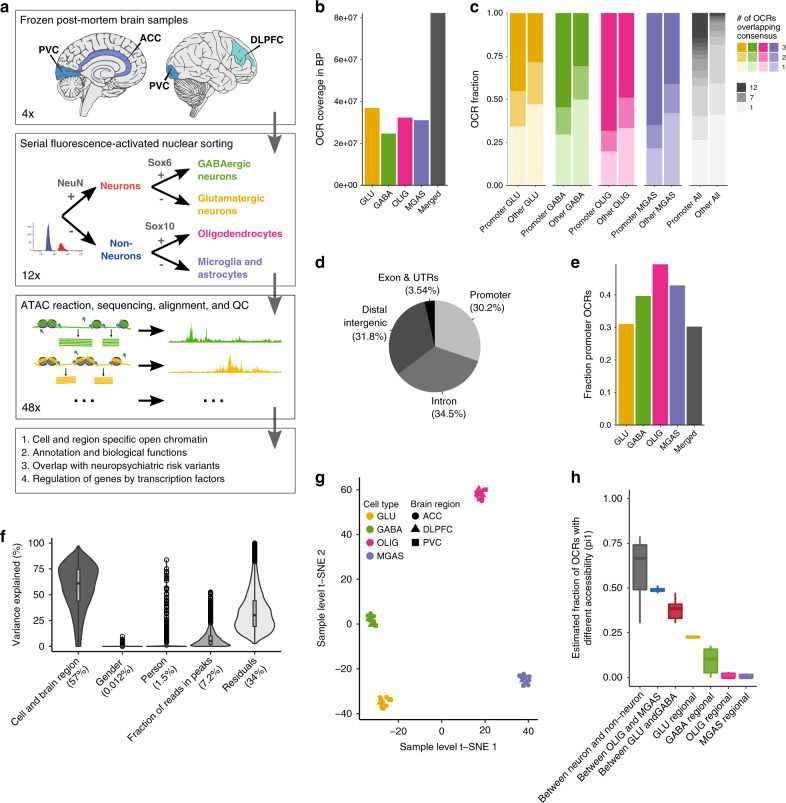
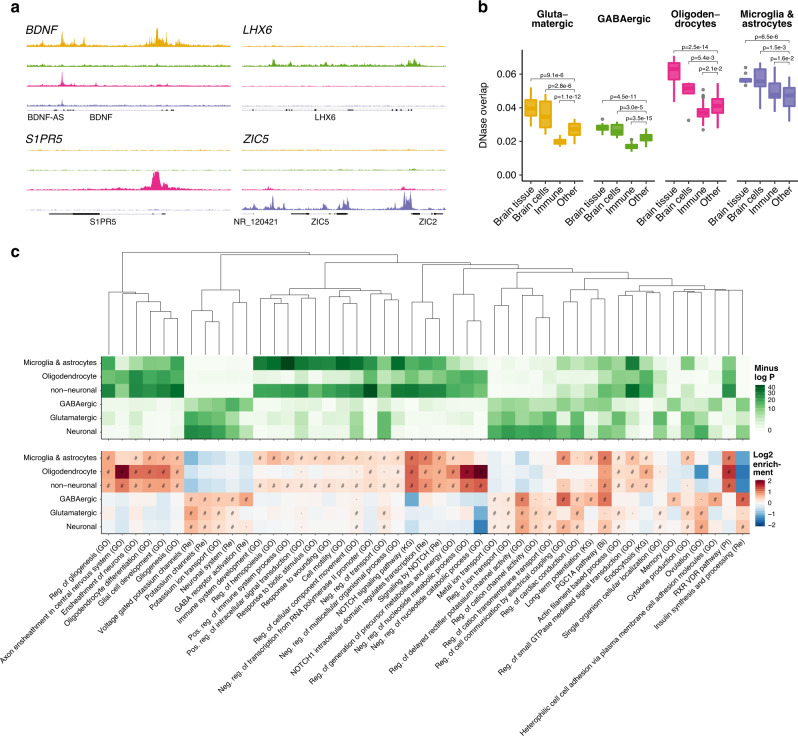
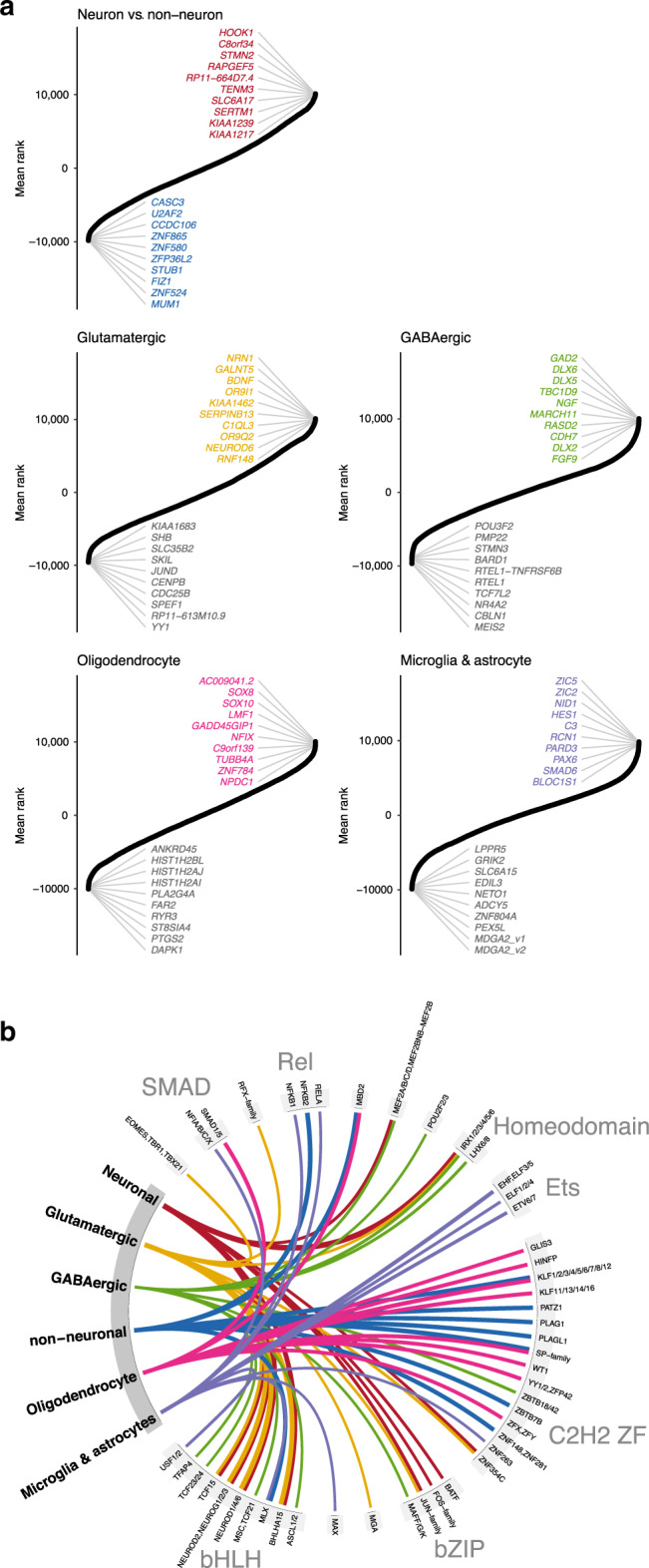
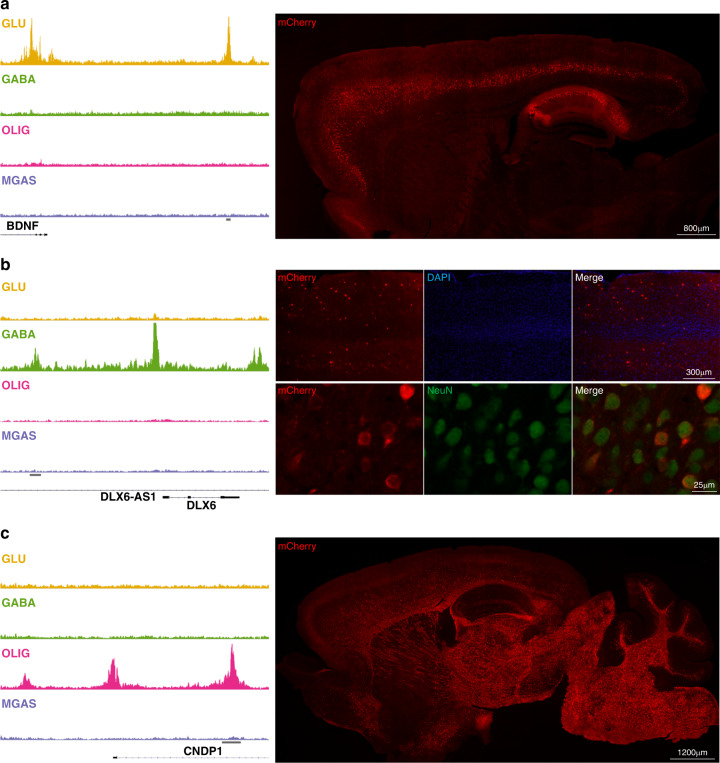
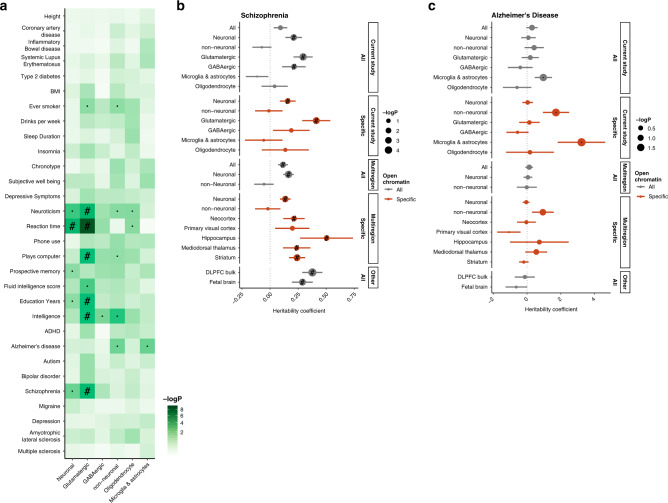
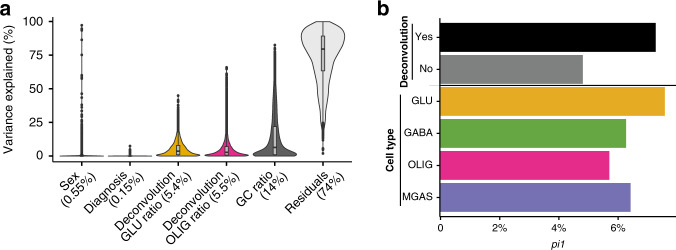
The chromatin landscape of human brain cells encompasses key information to understanding brain function. Here we use ATAC-seq to profile the chromatin structure in four distinct populations of cells (glutamatergic neurons, GABAergic neurons, oligodendrocytes, and microglia/astrocytes) from three different brain regions (anterior cingulate cortex, dorsolateral prefrontal cortex, and primary visual cortex) in human postmortem brain samples. We find that chromatin accessibility varies greatly by cell type and, more moderately, by brain region, with glutamatergic neurons showing the largest regional variability. Transcription factor footprinting implicates cell-specific transcriptional regulators and infers cell-specific regulation of protein-coding genes, long intergenic noncoding RNAs and microRNAs. In vivo transgenic mouse experiments validate the cell type specificity of several of these human-derived regulatory sequences. We find that open chromatin regions in glutamatergic neurons are enriched for neuropsychiatric risk variants, particularly those associated with schizophrenia. Integration of cell-specific chromatin data with a bulk tissue study of schizophrenia brains increases statistical power and confirms that glutamatergic neurons are most affected. These findings illustrate the utility of studying the cell-type-specific epigenome in complex tissues like the human brain, and the potential of such approaches to better understand the genetic basis of human brain function.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
