

Association of genetic mutations and loss of ambulation in childhood-onset dystrophinopathy
- PMID: 33150975
- PMCID: PMC8094042
- DOI: 10.1002/mus.27113
Association of genetic mutations and loss of ambulation in childhood-onset dystrophinopathy
Abstract
Background: Quantifying associations between genetic mutations and loss of ambulation (LoA) among males diagnosed with childhood-onset dystrophinopathy is important for understanding variation in disease progression and may be useful in clinical trial design.
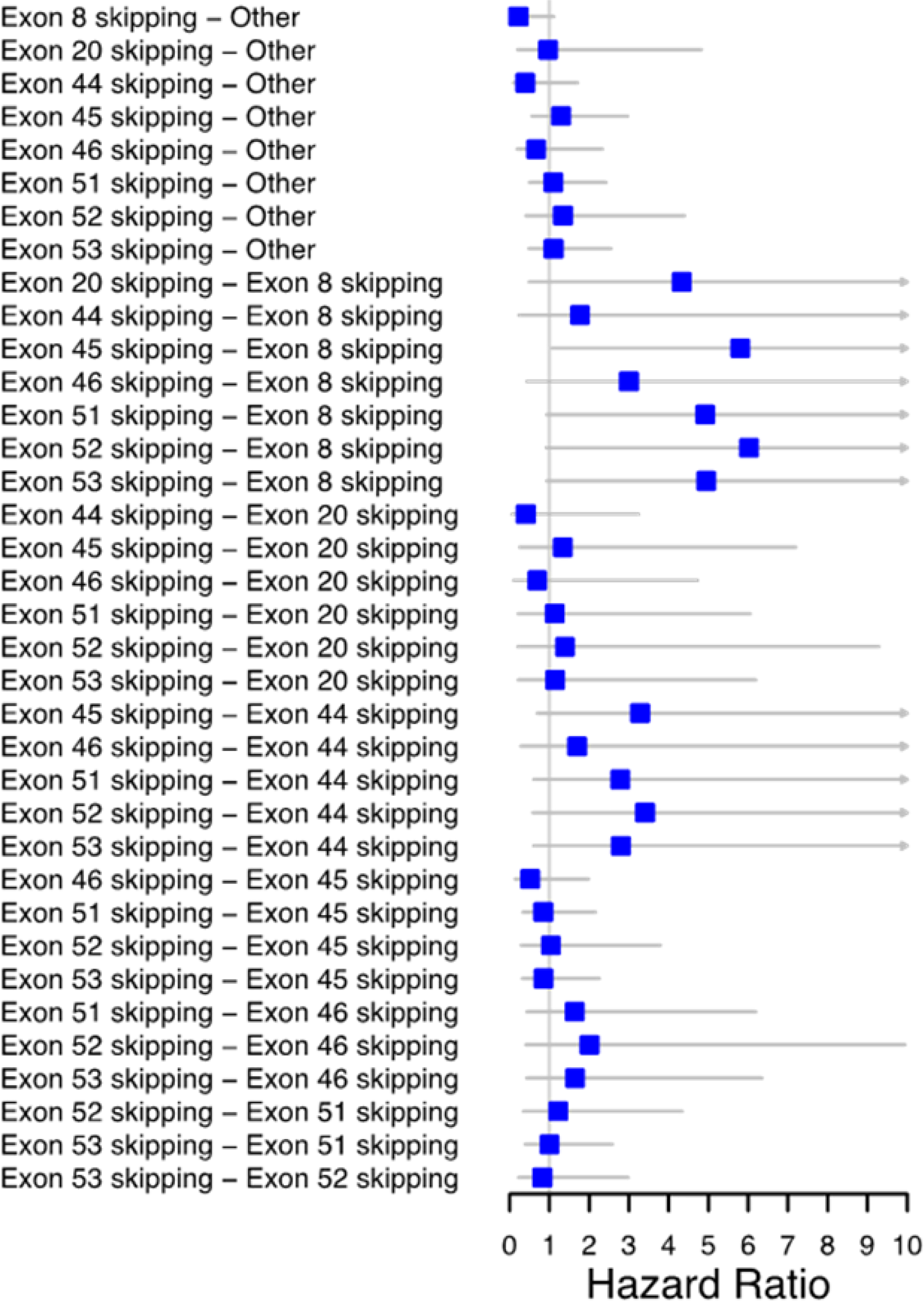
Methods: Genetic and clinical data from the Muscular Dystrophy Surveillance, Tracking, and Research Network for 358 males born and diagnosed from 1982 to 2011 were analyzed. LoA was defined as the age at which independent ambulation ceased. Genetic mutations were defined by overall type (deletion/duplication/point mutation) and among deletions, those amenable to exon-skipping therapy (exons 8, 20, 44-46, 51-53) and another group. Cox proportional hazards regression modeling was used to estimate hazard ratios (HRs) and 95% confidence intervals (CIs).
Results: Mutation type did not predict time to LoA. Controlling for corticosteroids, Exons 8 (HR = 0.22; 95% CI = 0.08, 0.63) and 44 (HR = 0.30; 95% CI = 0.12, 0.78) were associated with delayed LoA compared to other exon deletions.
Conclusions: Delayed LoA in males with mutations amenable to exon-skipping therapy is consistent with previous studies. These findings suggest that clinical trials including exon 8 and 44 skippable males should consider mutation information prior to randomization.
Keywords: Duchenne muscular dystrophy; MD STARnet; exon skipping; loss of ambulation; natural history study.
© 2020 Wiley Periodicals LLC.
Figures


References
-
- Kunkel LM, Hejtmancik JF, Caskey CT, et al. Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature. 1986;322(6074):73–77. - PubMed
-
- Magri F, Govoni A, D’Angelo MG, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258(9):1610–1623. - PubMed
-
- Humbertclaude V, Hamroun D, Bezzou K, et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. European Journal of Paediatric Neurology. 2012;16(2):149–160. - PubMed
-
- Goemans N, van den Hauwe M, Wilson R, van Impe A, Klingels K, Buyse G. Ambulatory capacity and disease progression as measured by the 6-minute-walk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscular Disorders. 2013;23(8):618–623. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 NS097631/NS/NINDS NIH HHS/United States
- U01 DD000187/DD/NCBDD CDC HHS/United States
- U01DD001108/ACL/ACL HHS/United States
- R01 FD006071/FD/FDA HHS/United States
- U01 DD000191/DD/NCBDD CDC HHS/United States
- CC999999/ImCDC/Intramural CDC HHS/United States
- U01 DD001117/DD/NCBDD CDC HHS/United States
- U01 DD000392/DD/NCBDD CDC HHS/United States
- U01 DD001126/DD/NCBDD CDC HHS/United States
- U01 DD001123/DD/NCBDD CDC HHS/United States
- R01 NS104010/NS/NINDS NIH HHS/United States
- U01DD001126/ACL/ACL HHS/United States
- U01 DD001246/DD/NCBDD CDC HHS/United States
- U01 DD000189/DD/NCBDD CDC HHS/United States
- U01 DD001116/DD/NCBDD CDC HHS/United States
- U01DD001116/ACL/ACL HHS/United States
- U01 DD001247/DD/NCBDD CDC HHS/United States
- U01DD001119/ACL/ACL HHS/United States
- U01DD001123/ACL/ACL HHS/United States
- K23 NS091511/NS/NINDS NIH HHS/United States
- U01 DD001249/DD/NCBDD CDC HHS/United States
- U01 DD001119/DD/NCBDD CDC HHS/United States
- U01 DD001108/DD/NCBDD CDC HHS/United States
- U01 DD001248/DD/NCBDD CDC HHS/United States
- U01DD001117/ACL/ACL HHS/United States
- U01 DD001255/DD/NCBDD CDC HHS/United States
- U01 DD000190/DD/NCBDD CDC HHS/United States
LinkOut - more resources
Full Text Sources
