

TSGIT: An N- and C-terminal tandem tag system for purification of native and intein-mediated ligation-ready proteins
- PMID: 33150985
- PMCID: PMC7784762
- DOI: 10.1002/pro.3989
TSGIT: An N- and C-terminal tandem tag system for purification of native and intein-mediated ligation-ready proteins
Abstract
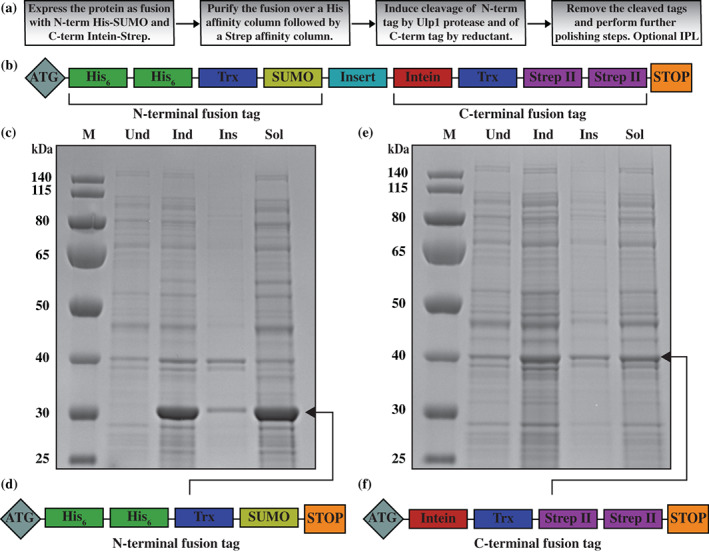
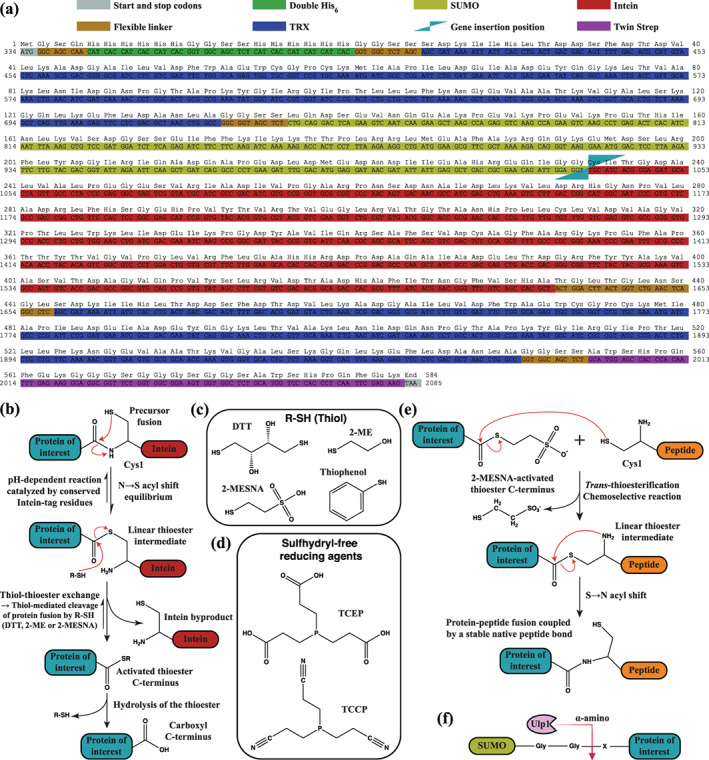
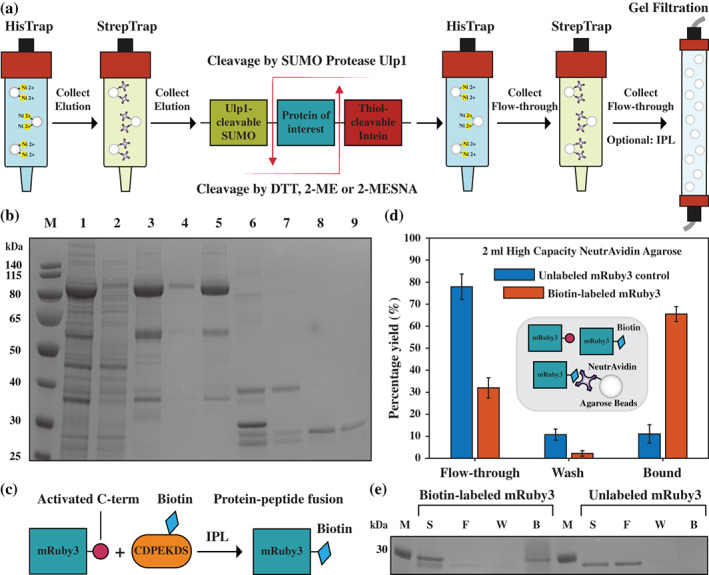
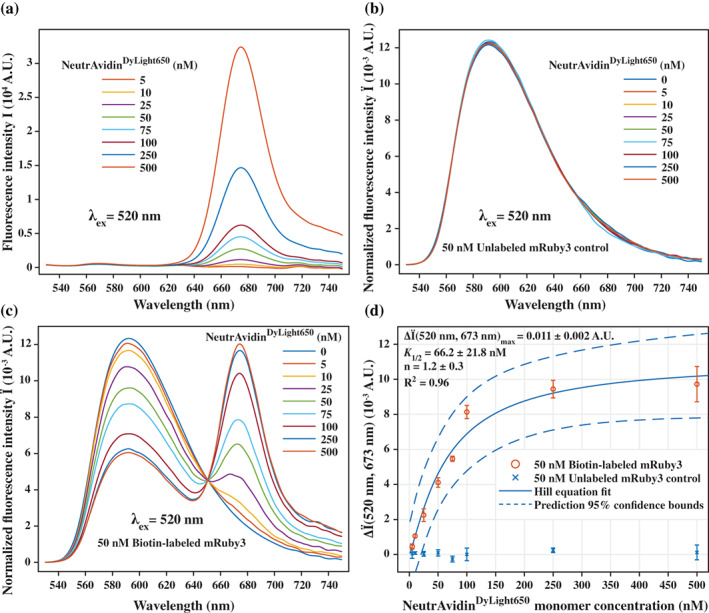
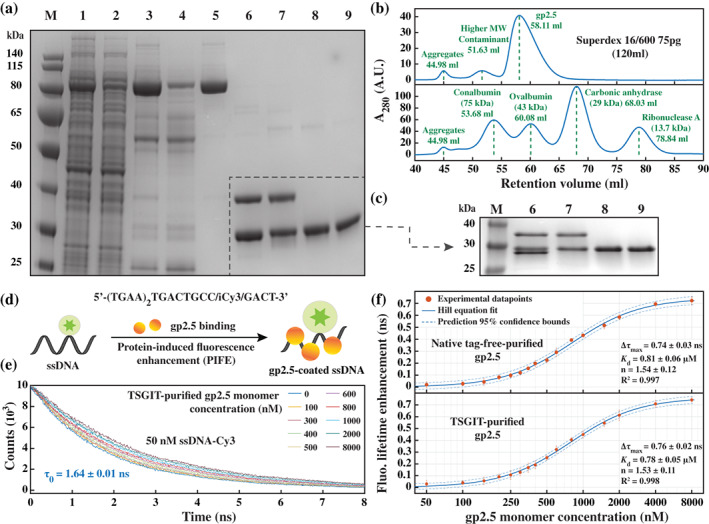
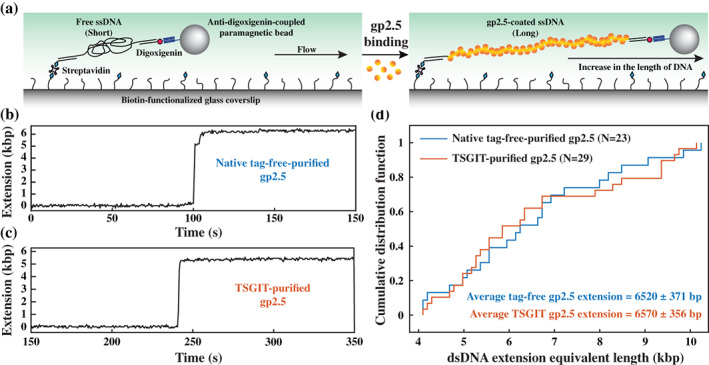
A large variety of fusion tags have been developed to improve protein expression, solubilization, and purification. Nevertheless, these tags have been combined in a rather limited number of composite tags and usually these composite tags have been dictated by traditional commercially-available expression vectors. Moreover, most commercially-available expression vectors include either N- or C-terminal fusion tags but not both. Here, we introduce TSGIT, a fusion-tag system composed of both N- and a C-terminal composite fusion tags. The system includes two affinity tags, two solubilization tags and two cleavable tags distributed at both termini of the protein of interest. Therefore, the N- and the C-terminal composite fusion tags in TSGIT are fully orthogonal in terms of both affinity selection and cleavage. For using TSGIT, we streamlined the cloning, expression, and purification procedures. Each component tag is selected to maximize its benefits toward the final construct. By expressing and partially purifying the protein of interest between the components of the TSGIT fusion, the full-length protein is selected over truncated forms, which has been a long-standing problem in protein purification. Moreover, due to the nature of the cleavable tags in TSGIT, the protein of interest is obtained in its native form without any additional undesired N- or C-terminal amino acids. Finally, the resulting purified protein is ready for efficient ligation with other proteins or peptides for downstream applications. We demonstrate the use of this system by purifying a large amount of native fluorescent mRuby3 protein and bacteriophage T7 gp2.5 ssDNA-binding protein.
Keywords: IPL; Intein; SUMO; biotin; fusion tag; protein cleavage; protein degradation; protein expression; protein ligation; purification tag; truncated protein.
© 2020 The Authors. Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Terpe K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. - PubMed
-
- Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol J. 2012;7:620–634. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
