

Cleavable Cross-Linkers and Mass Spectrometry for the Ultimate Task of Profiling Protein-Protein Interaction Networks in Vivo
- PMID: 33151691
- PMCID: PMC7786381
- DOI: 10.1021/acs.jproteome.0c00583
Cleavable Cross-Linkers and Mass Spectrometry for the Ultimate Task of Profiling Protein-Protein Interaction Networks in Vivo
Abstract
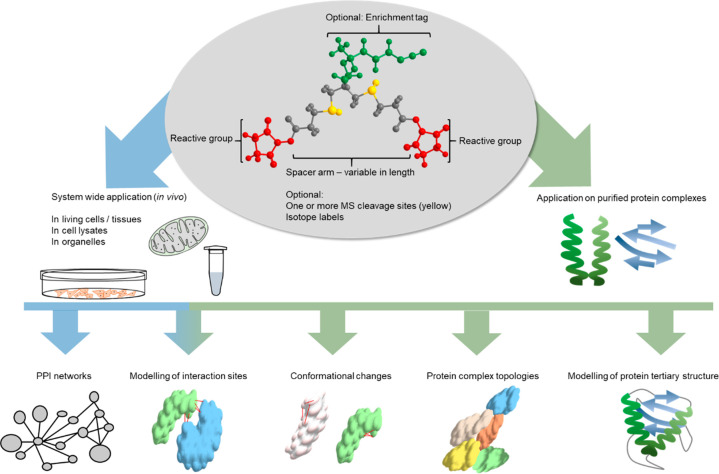
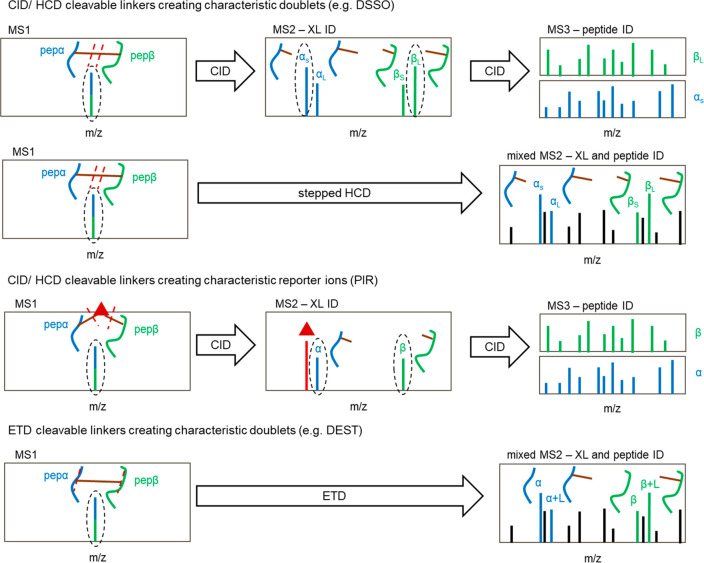
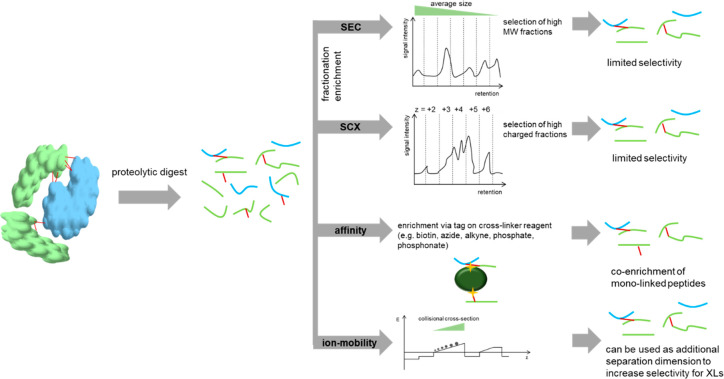
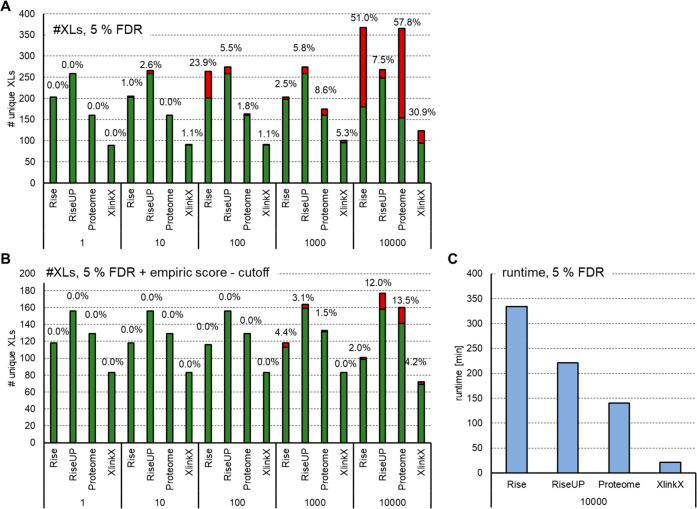
Cross-linking mass spectrometry (XL-MS) has matured into a potent tool to identify protein-protein interactions or to uncover protein structures in living cells, tissues, or organelles. The unique ability to investigate the interplay of proteins within their native environment delivers valuable complementary information to other advanced structural biology techniques. This Review gives a comprehensive overview of the current possible applications as well as the remaining limitations of the technique, focusing on cross-linking in highly complex biological systems like cells, organelles, or tissues. Thanks to the commercial availability of most reagents and advances in user-friendly data analysis, validation, and visualization tools, studies using XL-MS can, in theory, now also be utilized by nonexpert laboratories.
Keywords: acquisition techniques; cross-linking; fragmentation techniques; guideline; mass spectrometry.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
