

Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms
- PMID: 33151911
- PMCID: PMC7810480
- DOI: 10.1172/JCI95747
Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms
Abstract
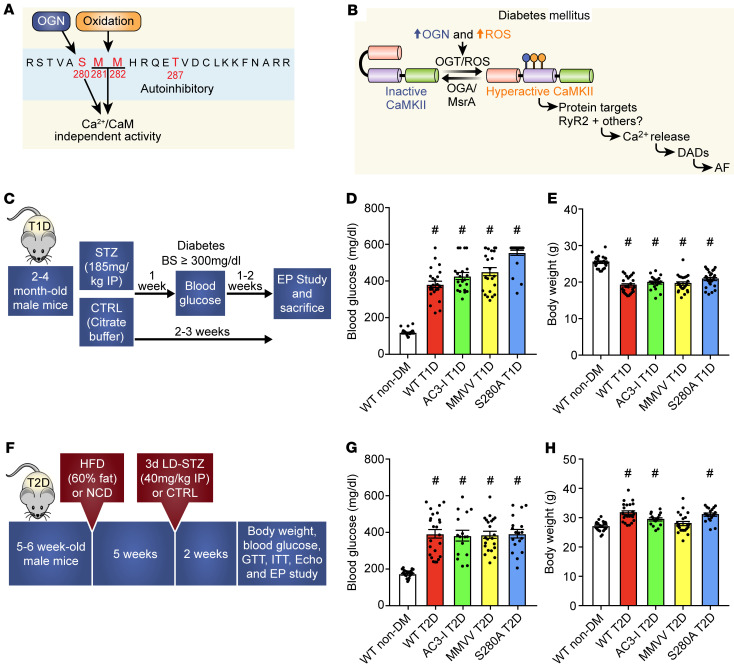
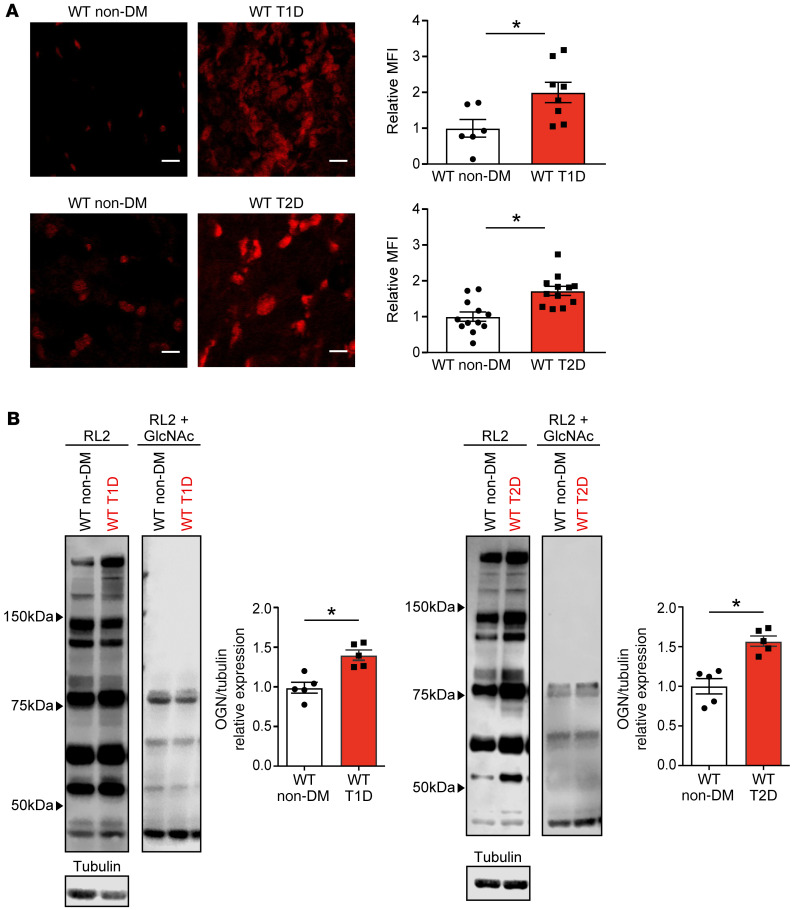
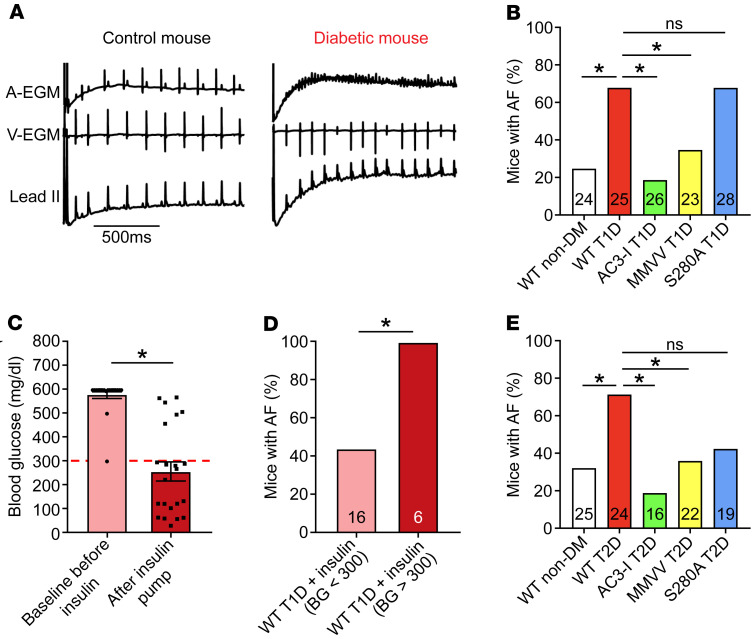
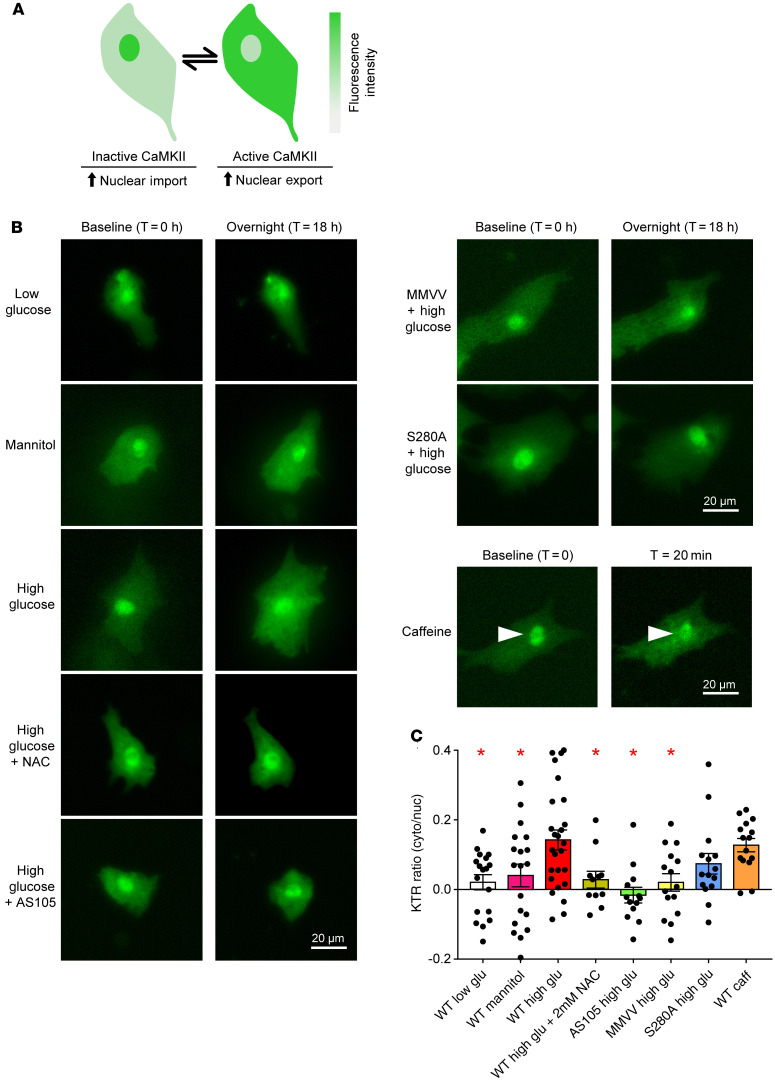
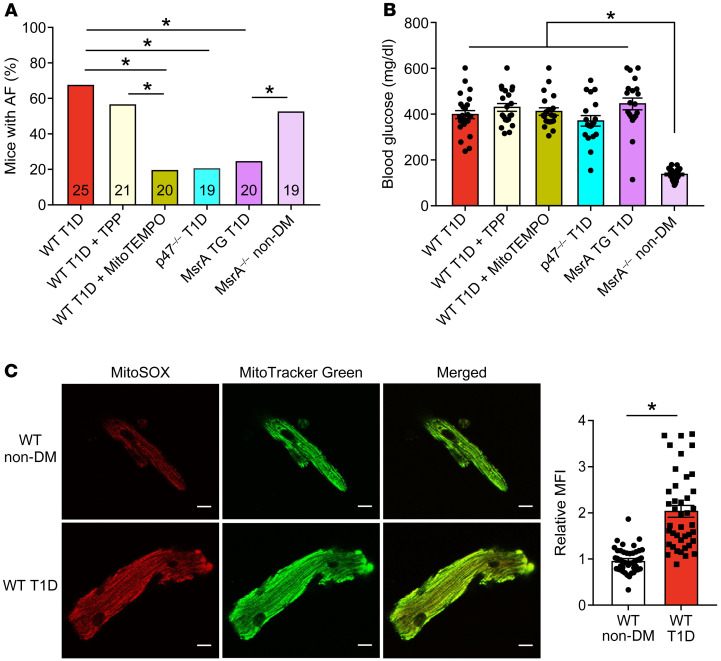
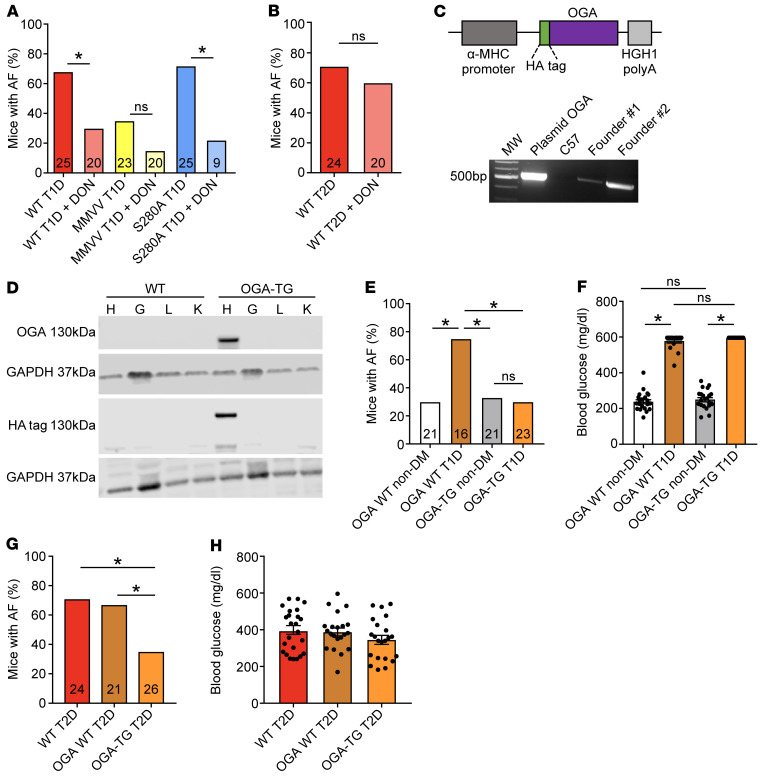
Diabetes mellitus (DM) and atrial fibrillation (AF) are major unsolved public health problems, and diabetes is an independent risk factor for AF. However, the mechanism(s) underlying this clinical association is unknown. ROS and protein O-GlcNAcylation (OGN) are increased in diabetic hearts, and calmodulin kinase II (CaMKII) is a proarrhythmic signal that may be activated by ROS (oxidized CaMKII, ox-CaMKII) and OGN (OGN-CaMKII). We induced type 1 (T1D) and type 2 DM (T2D) in a portfolio of genetic mouse models capable of dissecting the role of ROS and OGN at CaMKII and global OGN in diabetic AF. Here, we showed that T1D and T2D significantly increased AF, and this increase required CaMKII and OGN. T1D and T2D both required ox-CaMKII to increase AF; however, we did not detect OGN-CaMKII or a role for OGN-CaMKII in diabetic AF. Collectively, our data affirm CaMKII as a critical proarrhythmic signal in diabetic AF and suggest ROS primarily promotes AF by ox-CaMKII, while OGN promotes AF by a CaMKII-independent mechanism(s). These results provide insights into the mechanisms for increased AF in DM and suggest potential benefits for future CaMKII and OGN targeted therapies.
Keywords: Arrhythmias; Cardiology; Diabetes; Protein kinases.
Conflict of interest statement
Figures
References
-
- Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. US Department of Health and Human Services; 2014. https://stacks.cdc.gov/view/cdc/23442/cdc_23442_DS1.pdf Accessed November 4, 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
