

Molecular screening of antimalarial, antiviral, anti-inflammatory and HIV protease inhibitors against spike glycoprotein of coronavirus
- PMID: 33152616
- PMCID: PMC7553127
- DOI: 10.1016/j.jmgm.2020.107769
Molecular screening of antimalarial, antiviral, anti-inflammatory and HIV protease inhibitors against spike glycoprotein of coronavirus
Abstract
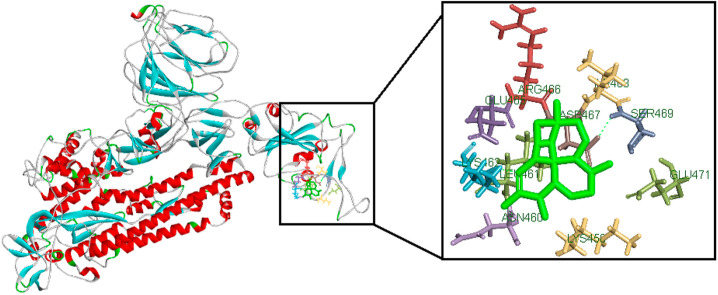
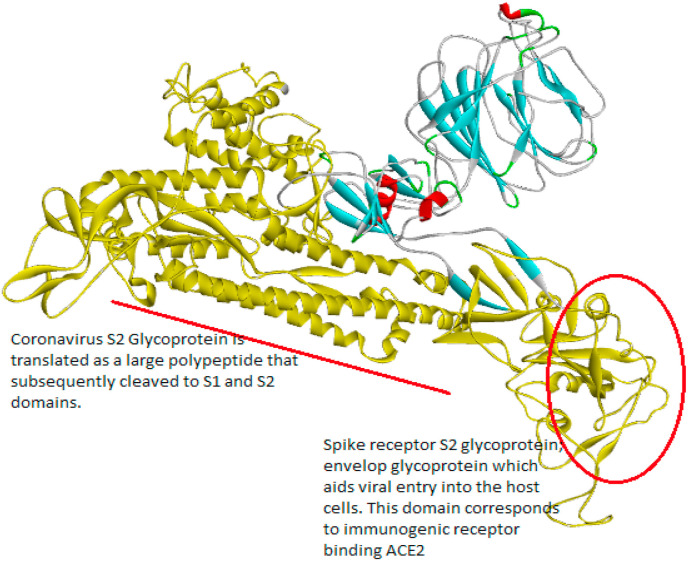
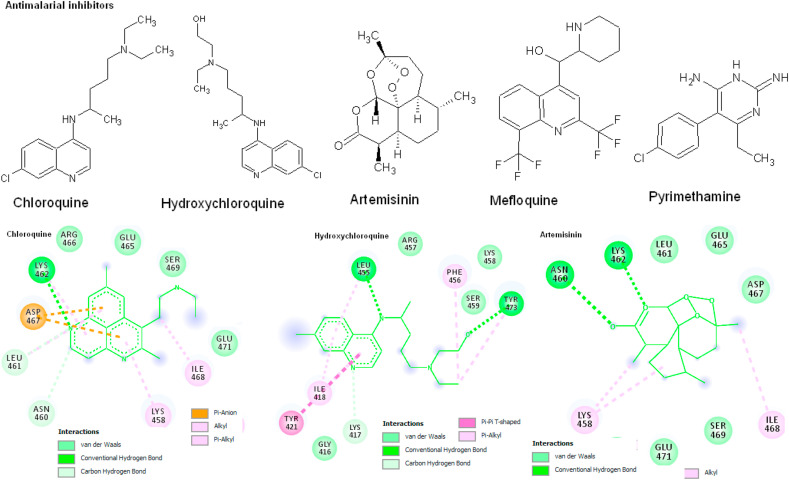
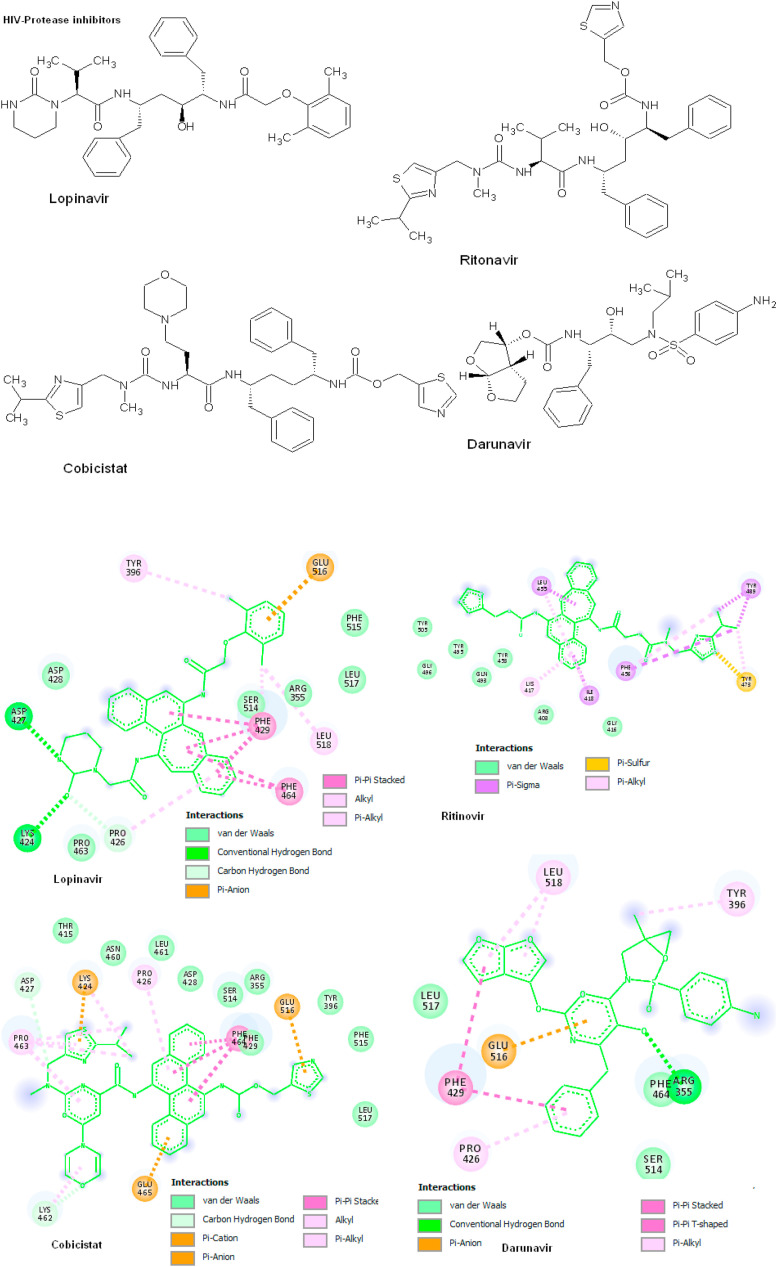
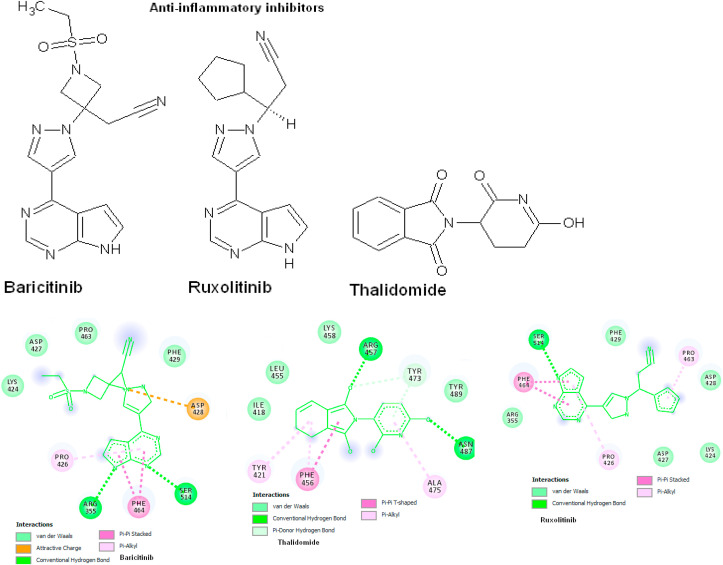
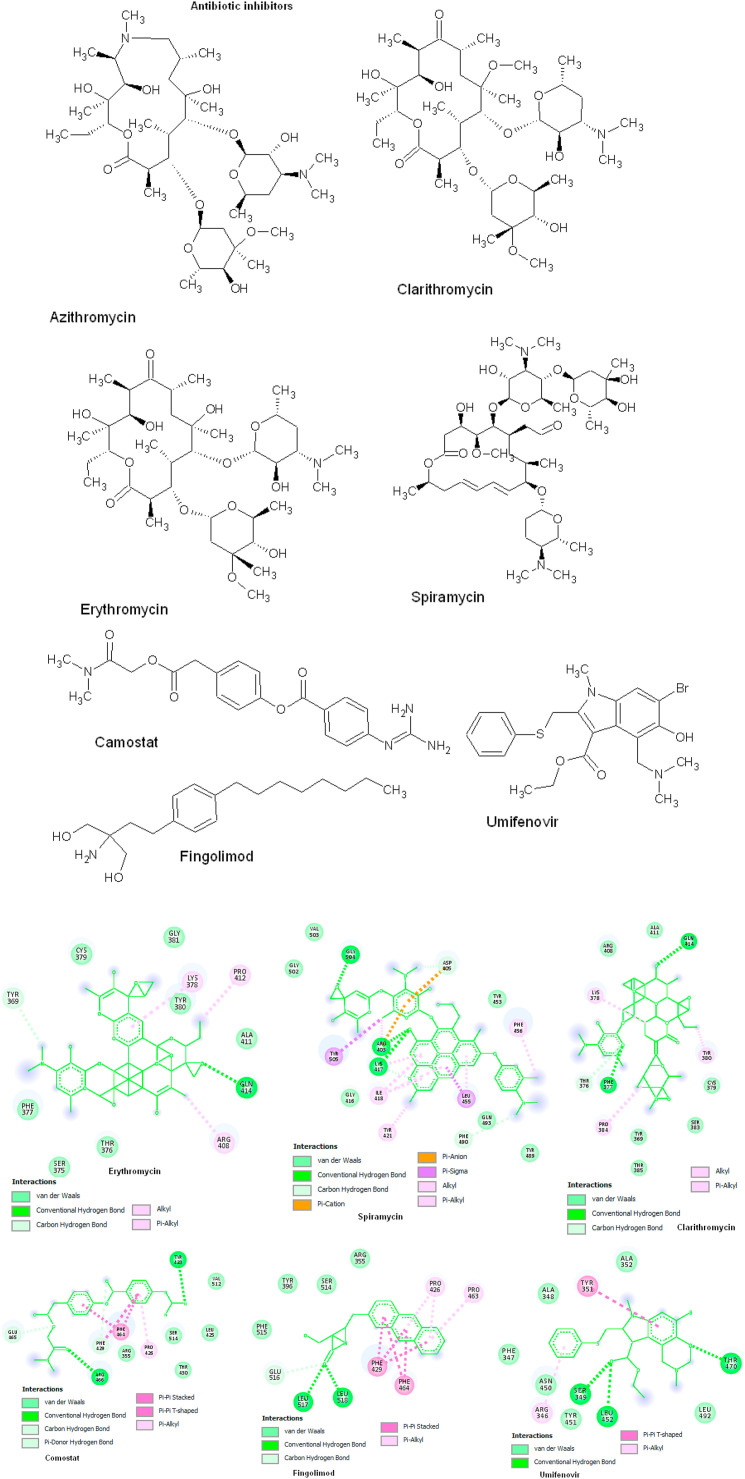
Coronavirus outbreak in December 2019 (COVID-19) is an emerging viral disease that poses major menace to Humans and it's a crucial need to find the possible treatment strategies. Spike protein (S2), a envelop glycoprotein aids viral entry into the host cells that corresponds to immunogenic ACE2 receptor binding and represents a potential antiviral drug target. Several drugs such as antimalarial, antibiotic, anti-inflammatory and HIV-protease inhibitors are currently undergoing treatment as clinical studies to test the efficacy and safety of COVID-19. Some promising results have been observed with the patients and also with high mortality rate. Hence, there is a need to screen the best CoV inhibitors using insilico analysis. The Molecular methodologies applied in the present study are, Molecular docking, virtual screening, drug-like and ADMET prediction helps to target CoV inhibitors. The results were screened based on docking score, H-bonds, and amino acid interactions. The results shows HIV-protease inhibitors such as cobicistat (-8.3kcal/mol), Darunavir (-7.4kcal/mol), Lopinavir (-9.1kcal/mol) and Ritonavir (-8.0 kcal/mol), anti-inflammatory drugs such as Baricitinib (-5.8kcal/mol), Ruxolitinib (-6.5kcal/mol), Thalidomide (-6.5kcal/mol), antibiotic drugs such as Erythromycin(-9.0kcal/mol) and Spiramycin (-8.5kcal/mol) molecules have good affinity towards spike protein compared to antimalarial drugs Chloroquine (-6.2kcal/mol), Hydroxychloroquine (-5.2kcal/mol) and Artemisinin (-6.8kcal/mol) have poor affinity to spike protein. The insilico pharmacological evaluation shows that these molecules exhibit good affinity of drug-like and ADMET properties. Hence, we propose that HIVprotease, anti-inflammatory and antibiotic inhibitors are the potential lead drug molecules for spike protein and preclinical studies needed to confirm the promising therapeutic ability against COVID-19.
Keywords: Anti-inflammatory drugs; Antiviral drugs; COVID-19; Coronavirus; Homology modeling; Molecular docking.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Molecular screening of glycyrrhizin-based inhibitors against ACE2 host receptor of SARS-CoV-2.J Mol Model. 2021 Jun 24;27(7):206. doi: 10.1007/s00894-021-04816-y. J Mol Model. 2021. PMID: 34169390 Free PMC article.
-
In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein-HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs.Viruses. 2024 Oct 31;16(11):1726. doi: 10.3390/v16111726. Viruses. 2024. PMID: 39599841 Free PMC article.
-
In silico Potential of Approved Antimalarial Drugs for Repurposing Against COVID-19.OMICS. 2020 Oct;24(10):568-580. doi: 10.1089/omi.2020.0071. Epub 2020 Jul 30. OMICS. 2020. PMID: 32757981
-
Structure based Drug Designing Approaches in SARS-CoV-2 Spike Inhibitor Design.Curr Top Med Chem. 2022;22(29):2396-2409. doi: 10.2174/1568026623666221103091658. Curr Top Med Chem. 2022. PMID: 36330617 Review.
-
Computational Lock and Key and Dynamic Trajectory Analysis of Natural Biophors Against COVID-19 Spike Protein to Identify Effective Lead Molecules.Mol Biotechnol. 2021 Oct;63(10):898-908. doi: 10.1007/s12033-021-00358-z. Epub 2021 Jun 22. Mol Biotechnol. 2021. PMID: 34159564 Free PMC article. Review.
Cited by
-
Advanced Drug Delivery Platforms for the Treatment of Oral Pathogens.Pharmaceutics. 2022 Oct 26;14(11):2293. doi: 10.3390/pharmaceutics14112293. Pharmaceutics. 2022. PMID: 36365112 Free PMC article. Review.
-
Exploration of NMI-MsCl mediated amide bond formation for the synthesis of novel 3,5-substituted-1,2,4-oxadiazole derivatives: synthesis, evaluation of anti-inflammatory activity and molecular docking studies.Mol Divers. 2023 Aug;27(4):1867-1878. doi: 10.1007/s11030-022-10536-z. Epub 2022 Oct 11. Mol Divers. 2023. PMID: 36219380
-
Artemisia Extracts and Artemisinin-Based Antimalarials for COVID-19 Management: Could These Be Effective Antivirals for COVID-19 Treatment?Molecules. 2022 Jun 14;27(12):3828. doi: 10.3390/molecules27123828. Molecules. 2022. Retraction in: Molecules. 2023 Oct 30;28(21):7337. doi: 10.3390/molecules28217337. PMID: 35744958 Free PMC article. Retracted. Review.
-
SARS-CoV-2 Infection may be Prevented with Cytochrome Inhibitors: Cobicistat and Ritonavir.Infect Dis Clin Microbiol. 2022 Sep 26;4(3):185-191. doi: 10.36519/idcm.2022.139. eCollection 2022 Sep. Infect Dis Clin Microbiol. 2022. PMID: 38633393 Free PMC article.
-
An overview of the anti-SARS-CoV-2 properties of Artemisia annua, its antiviral action, protein-associated mechanisms, and repurposing for COVID-19 treatment.J Integr Med. 2021 Sep;19(5):375-388. doi: 10.1016/j.joim.2021.07.003. Epub 2021 Jul 22. J Integr Med. 2021. PMID: 34479848 Free PMC article. Review.
References
-
- Burrell Christopher J., Howard Colin R., Murphy Frederick A. fifth ed. Academic Press; 2017. Chapter 31 -Coronaviruses, Fenner and White’s Medical Virology; pp. 437–446.
-
- World Health Organization (WHO) WHO; China. Beijing: 2020. WHO Statement Regarding Cluster of Pneumonia Cases in Wuhan.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
