

A Small Molecule that Binds an RNA Repeat Expansion Stimulates Its Decay via the Exosome Complex
- PMID: 33157036
- PMCID: PMC7855261
- DOI: 10.1016/j.chembiol.2020.10.007
A Small Molecule that Binds an RNA Repeat Expansion Stimulates Its Decay via the Exosome Complex
Abstract
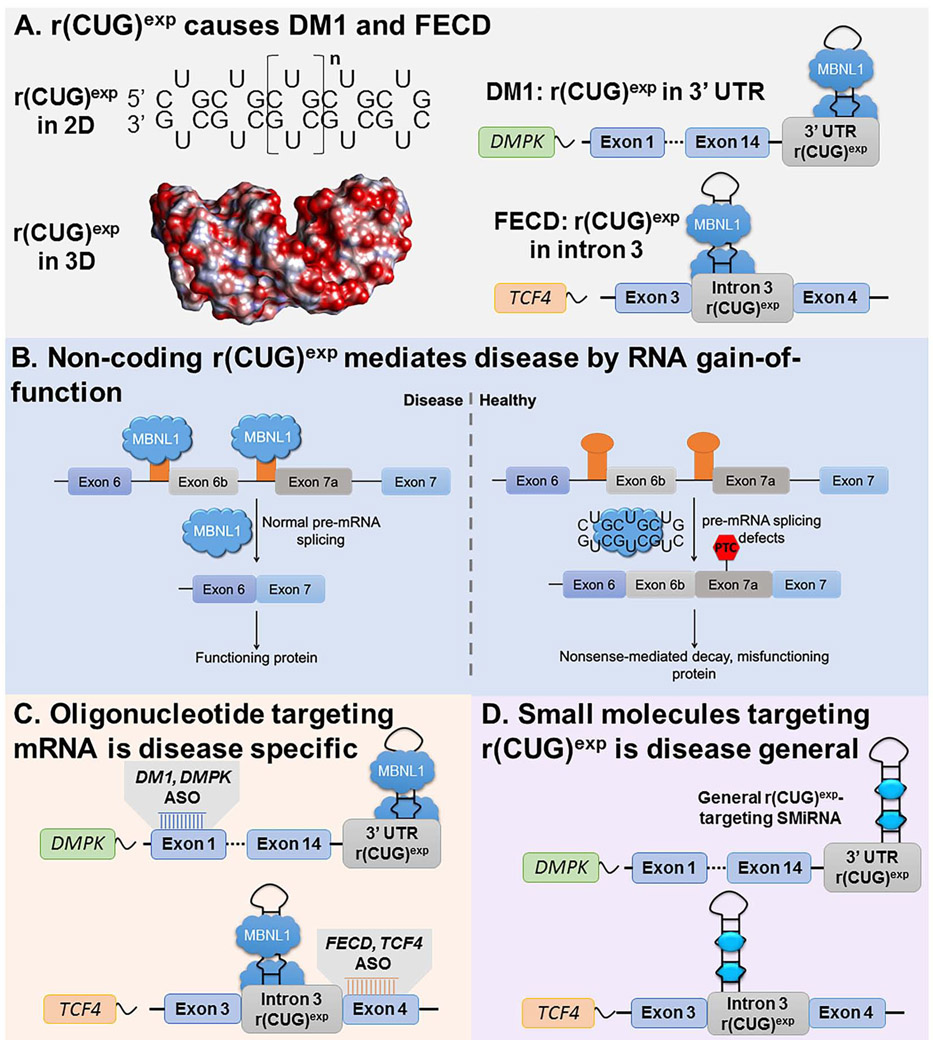
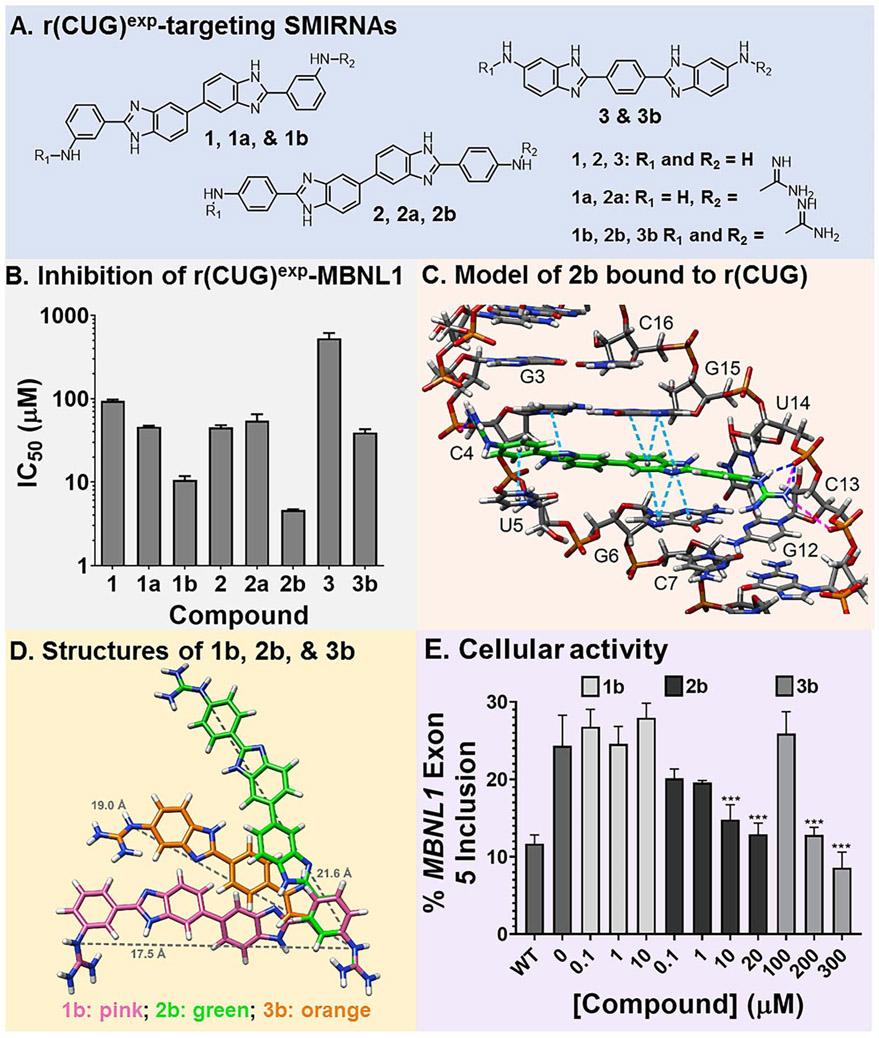
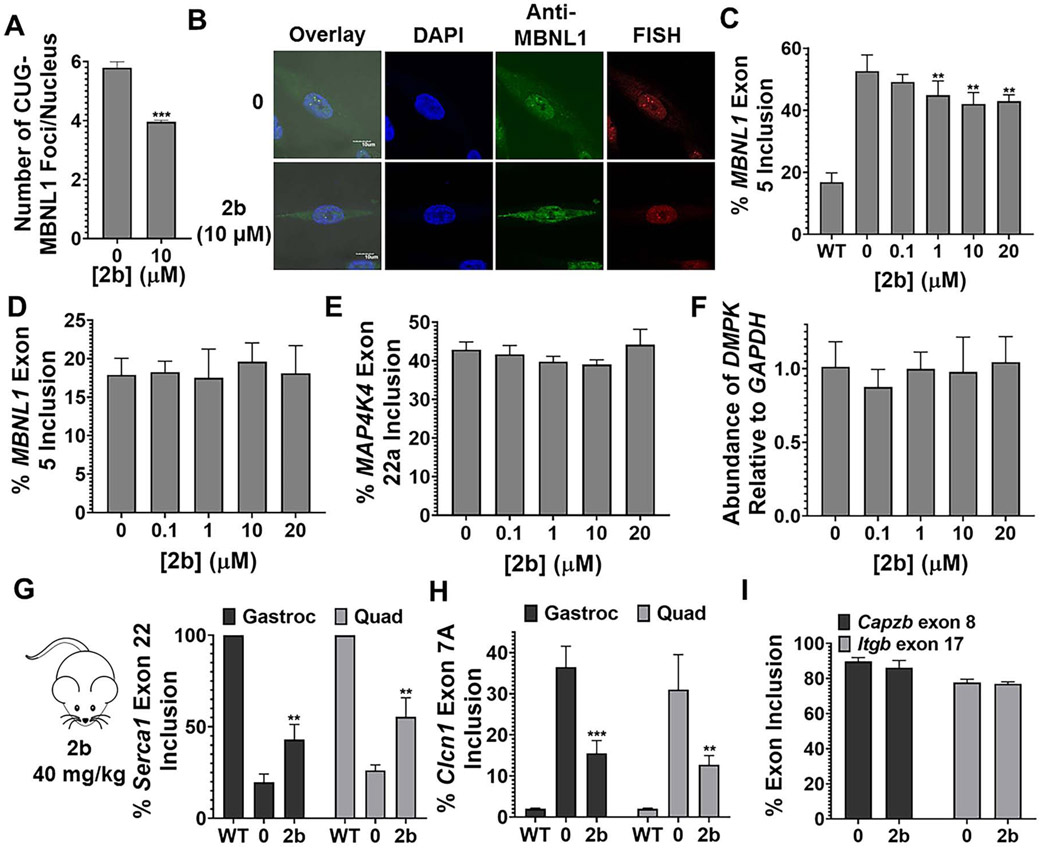
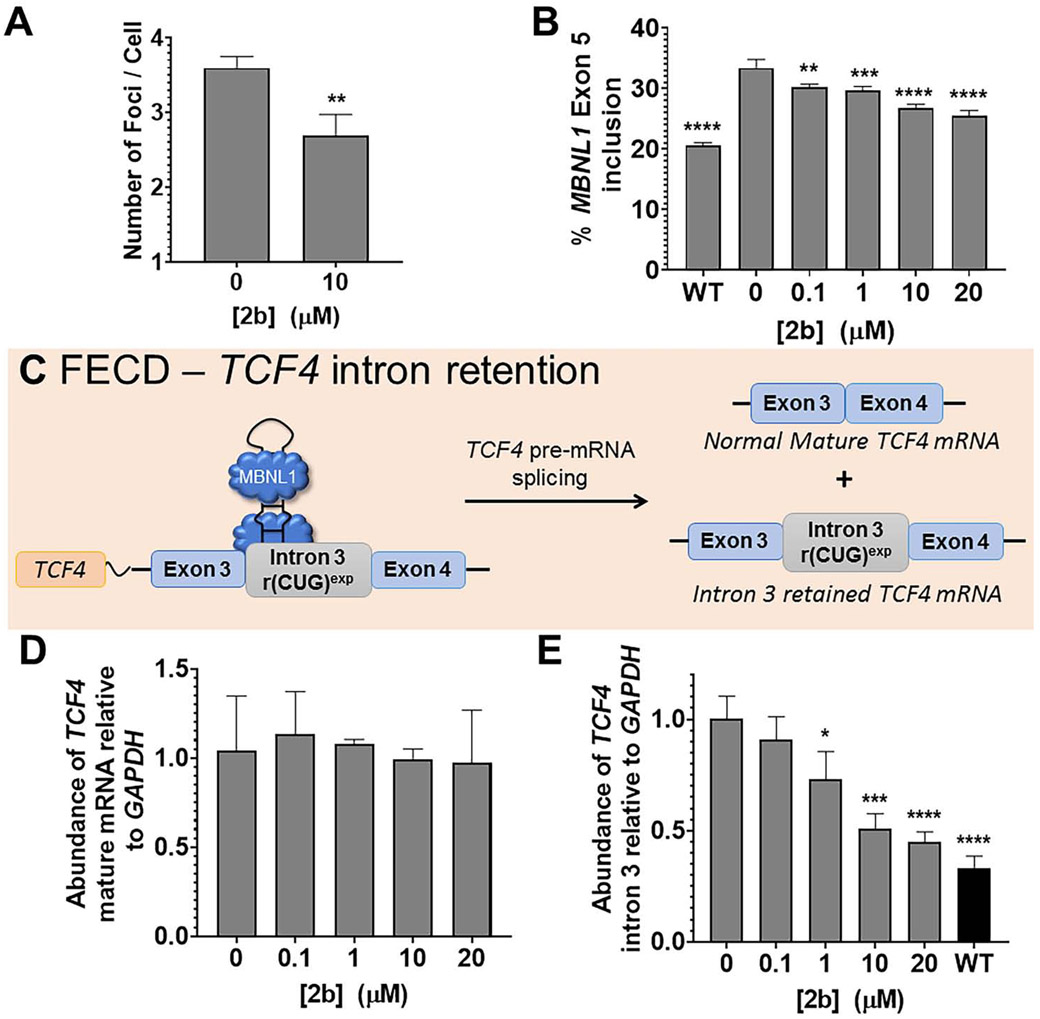
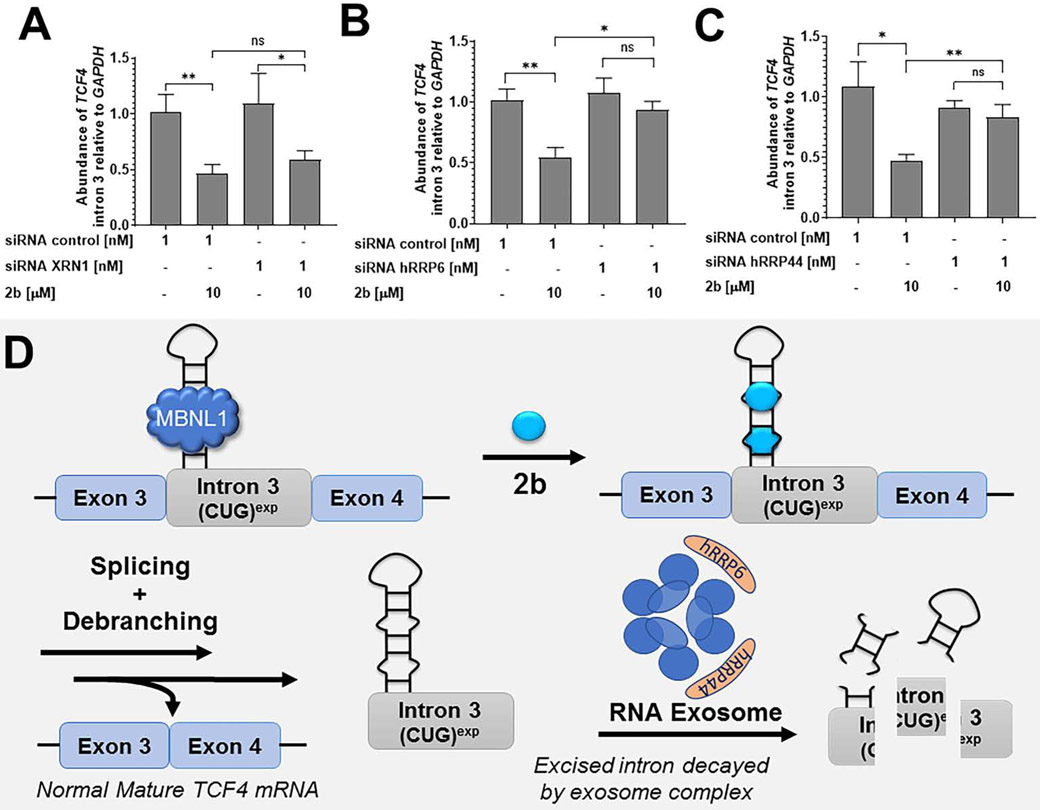
Many diseases are caused by toxic RNA repeats. Herein, we designed a lead small molecule that binds the structure of the r(CUG) repeat expansion [r(CUG)exp] that causes myotonic dystrophy type 1 (DM1) and Fuchs endothelial corneal dystrophy (FECD) and rescues disease biology in patient-derived cells and in vivo. Interestingly, the compound's downstream effects are different in the two diseases, owing to the location of the repeat expansion. In DM1, r(CUG)exp is harbored in the 3' untranslated region, and the compound has no effect on the mRNA's abundance. In FECD, however, r(CUG)exp is located in an intron, and the small molecule facilitates excision of the intron, which is then degraded by the RNA exosome complex. Thus, structure-specific, RNA-targeting small molecules can act disease specifically to affect biology, either by disabling the gain-of-function mechanism (DM1) or by stimulating quality control pathways to rid a disease-affected cell of a toxic RNA (FECD).
Keywords: RNA; RNA splicing; chemical biology; decay pathways; drug discovery; microsatellite disorders; targeted degradation.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests M.D.D. is a founder of Expansion Therapeutics, and S.G.R. and A.J.A. are currently employees of Expansion Therapeutics. M.D.D. and S.G.R. also have a patent related to this work (US20190152924A1).
Figures

Comment in
-
Small molecule targets toxic RNA repeats.Nat Rev Drug Discov. 2021 Jan;20(1):20. doi: 10.1038/d41573-020-00203-z. Nat Rev Drug Discov. 2021. PMID: 33173228 No abstract available.
References
-
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion J-P, Hudson T, et al. (1992). Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
