

Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2
- PMID: 33158276
- PMCID: PMC7672574
- DOI: 10.3390/ijms21218268
Coevolution, Dynamics and Allostery Conspire in Shaping Cooperative Binding and Signal Transmission of the SARS-CoV-2 Spike Protein with Human Angiotensin-Converting Enzyme 2
Abstract
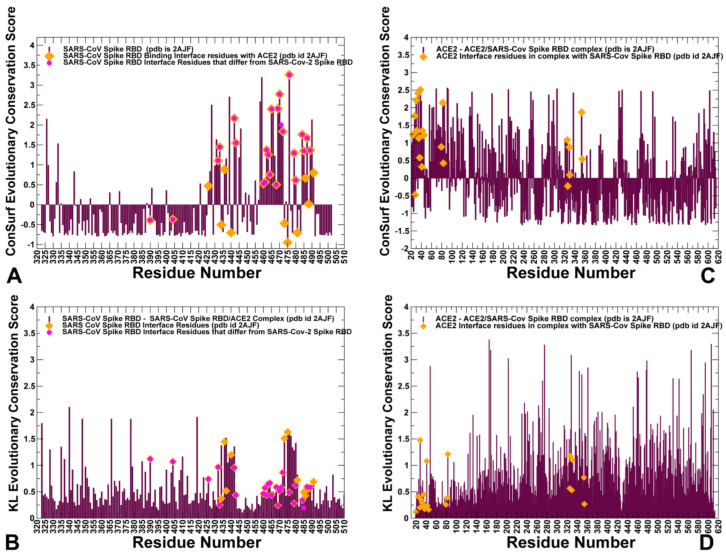
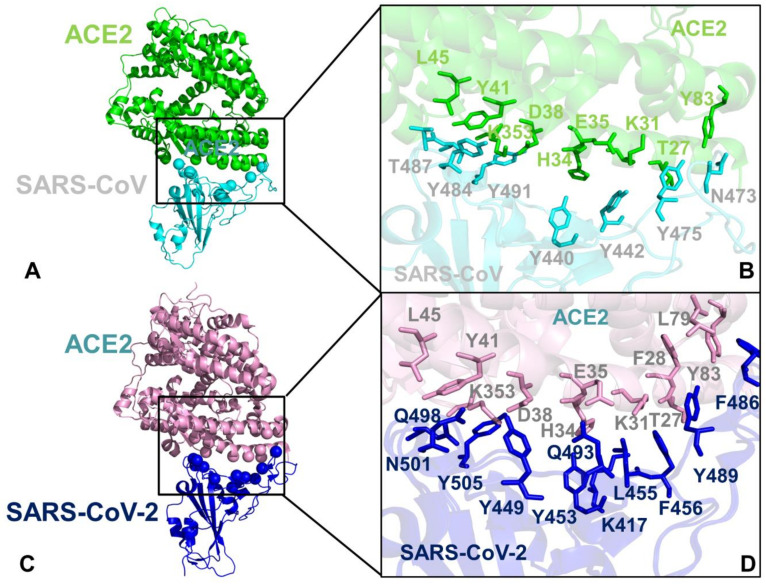
Binding to the host receptor is a critical initial step for the coronavirus SARS-CoV-2 spike protein to enter into target cells and trigger virus transmission. A detailed dynamic and energetic view of the binding mechanisms underlying virus entry is not fully understood and the consensus around the molecular origins behind binding preferences of SARS-CoV-2 for binding with the angiotensin-converting enzyme 2 (ACE2) host receptor is yet to be established. In this work, we performed a comprehensive computational investigation in which sequence analysis and modeling of coevolutionary networks are combined with atomistic molecular simulations and comparative binding free energy analysis of the SARS-CoV and SARS-CoV-2 spike protein receptor binding domains with the ACE2 host receptor. Different from other computational studies, we systematically examine the molecular and energetic determinants of the binding mechanisms between SARS-CoV-2 and ACE2 proteins through the lens of coevolution, conformational dynamics, and allosteric interactions that conspire to drive binding interactions and signal transmission. Conformational dynamics analysis revealed the important differences in mobility of the binding interfaces for the SARS-CoV-2 spike protein that are not confined to several binding hotspots, but instead are broadly distributed across many interface residues. Through coevolutionary network analysis and dynamics-based alanine scanning, we established linkages between the binding energy hotspots and potential regulators and carriers of signal communication in the virus-host receptor complexes. The results of this study detailed a binding mechanism in which the energetics of the SARS-CoV-2 association with ACE2 may be determined by cumulative changes of a number of residues distributed across the entire binding interface. The central findings of this study are consistent with structural and biochemical data and highlight drug discovery challenges of inhibiting large and adaptive protein-protein interfaces responsible for virus entry and infection transmission.
Keywords: ACE2; SARS-CoV spike protein; alanine scanning; allosteric interactions; binding free energy; coevolution; molecular dynamics; signal transmission.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
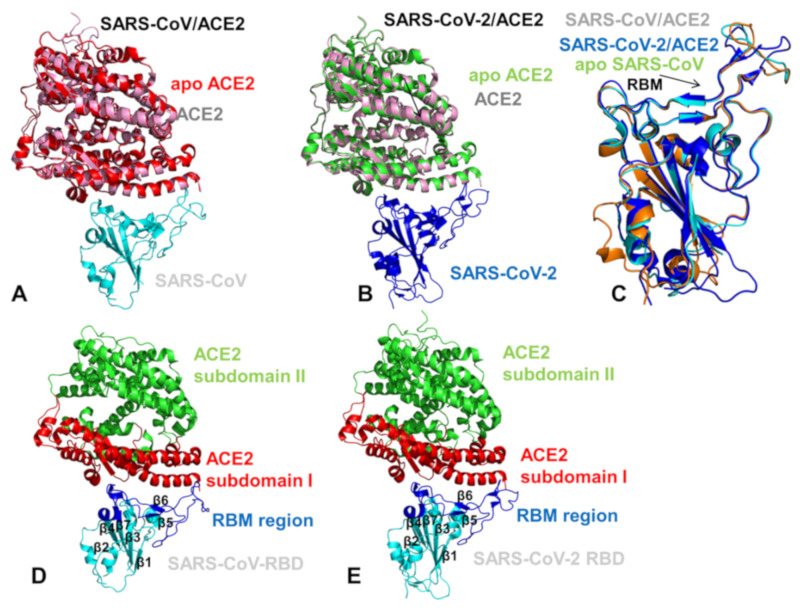
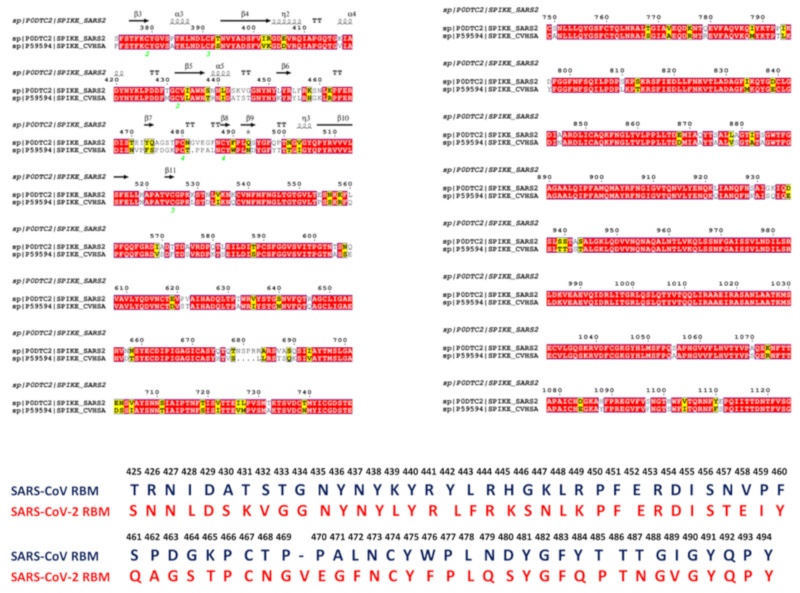
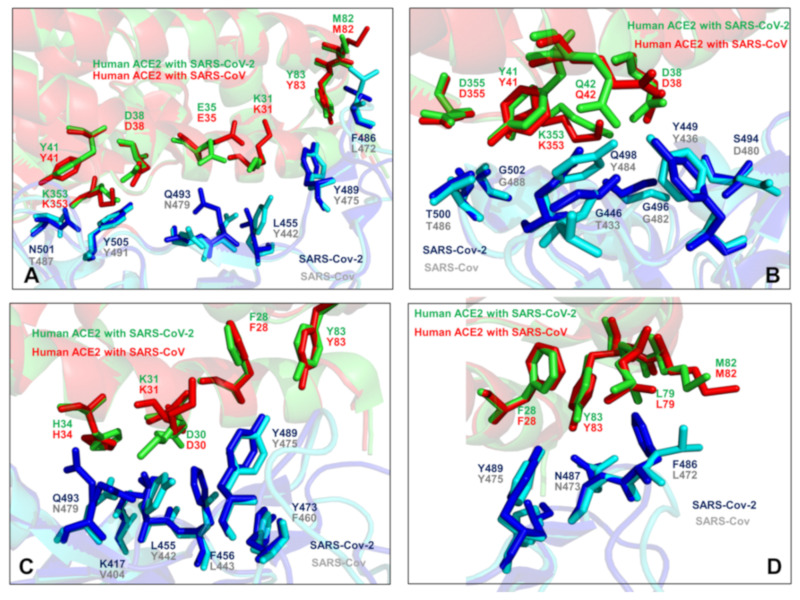
Similar articles
-
Computational Alanine Scanning and Structural Analysis of the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Complex.ACS Nano. 2020 Sep 22;14(9):11821-11830. doi: 10.1021/acsnano.0c04674. Epub 2020 Aug 26. ACS Nano. 2020. PMID: 32833435 Free PMC article.
-
Optimized Pseudotyping Conditions for the SARS-COV-2 Spike Glycoprotein.J Virol. 2020 Oct 14;94(21):e01062-20. doi: 10.1128/JVI.01062-20. Print 2020 Oct 14. J Virol. 2020. PMID: 32788194 Free PMC article.
-
Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites.ACS Nano. 2020 Aug 25;14(8):10616-10623. doi: 10.1021/acsnano.0c04798. Epub 2020 Aug 4. ACS Nano. 2020. PMID: 32806067
-
COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2.PLoS Pathog. 2020 Aug 21;16(8):e1008762. doi: 10.1371/journal.ppat.1008762. eCollection 2020 Aug. PLoS Pathog. 2020. PMID: 32822426 Free PMC article. Review.
-
Angiotensin-converting enzyme 2 (ACE2) receptor and SARS-CoV-2: Potential therapeutic targeting.Eur J Pharmacol. 2020 Oct 5;884:173455. doi: 10.1016/j.ejphar.2020.173455. Epub 2020 Jul 31. Eur J Pharmacol. 2020. PMID: 32745604 Free PMC article. Review.
Cited by
-
Mutational scanning of spike RBD protein for enhanced ACE2 affinity emerging Southeast Asia in the late transmission phase.Sci Rep. 2022 Apr 7;12(1):5896. doi: 10.1038/s41598-022-09999-9. Sci Rep. 2022. PMID: 35393512 Free PMC article.
-
Balancing Functional Tradeoffs between Protein Stability and ACE2 Binding in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB Lineages: Dynamics-Based Network Models Reveal Epistatic Effects Modulating Compensatory Dynamic and Energetic Changes.Viruses. 2023 May 10;15(5):1143. doi: 10.3390/v15051143. Viruses. 2023. PMID: 37243229 Free PMC article.
-
Insights into the mutation T1117I in the spike and the lineage B.1.1.389 of SARS-CoV-2 circulating in Costa Rica.Gene Rep. 2022 Jun;27:101554. doi: 10.1016/j.genrep.2022.101554. Epub 2022 Feb 8. Gene Rep. 2022. PMID: 35155843 Free PMC article.
-
E-Volve: understanding the impact of mutations in SARS-CoV-2 variants spike protein on antibodies and ACE2 affinity through patterns of chemical interactions at protein interfaces.PeerJ. 2022 Mar 22;10:e13099. doi: 10.7717/peerj.13099. eCollection 2022. PeerJ. 2022. PMID: 35341044 Free PMC article.
-
SARS-CoV-2 Genotyping Highlights the Challenges in Spike Protein Drift Independent of Other Essential Proteins.Microorganisms. 2024 Sep 9;12(9):1863. doi: 10.3390/microorganisms12091863. Microorganisms. 2024. PMID: 39338537 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
