

MEScan: a powerful statistical framework for genome-scale mutual exclusivity analysis of cancer mutations
- PMID: 33165532
- PMCID: PMC8189684
- DOI: 10.1093/bioinformatics/btaa957
MEScan: a powerful statistical framework for genome-scale mutual exclusivity analysis of cancer mutations
Abstract
Motivation: Cancer somatic driver mutations associated with genes within a pathway often show a mutually exclusive pattern across a cohort of patients. This mutually exclusive mutational signal has been frequently used to distinguish driver from passenger mutations and to investigate relationships among driver mutations. Current methods for de novo discovery of mutually exclusive mutational patterns are limited because the heterogeneity in background mutation rate can confound mutational patterns, and the presence of highly mutated genes can lead to spurious patterns. In addition, most methods only focus on a limited number of pre-selected genes and are unable to perform genome-wide analysis due to computational inefficiency.
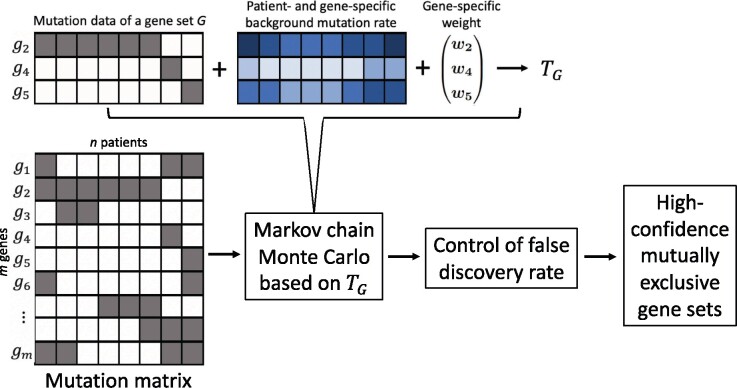
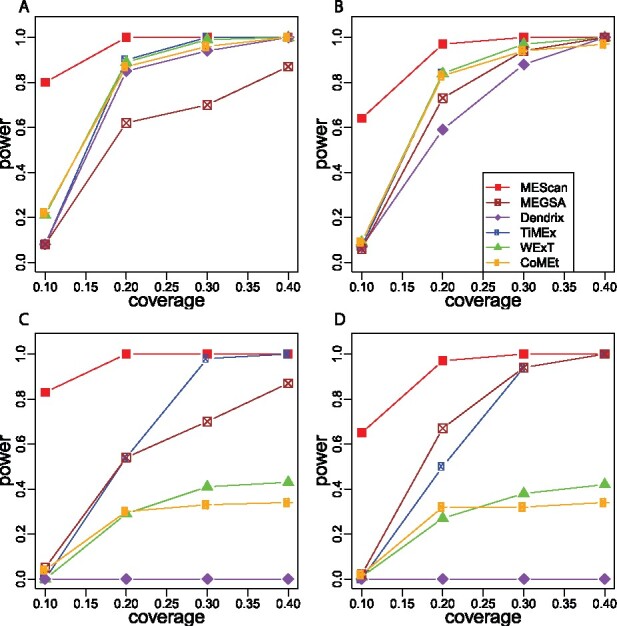
Results: We introduce a statistical framework, MEScan, for accurate and efficient mutual exclusivity analysis at the genomic scale. Our framework contains a fast and powerful statistical test for mutual exclusivity with adjustment of the background mutation rate and impact of highly mutated genes, and a multi-step procedure for genome-wide screening with the control of false discovery rate. We demonstrate that MEScan more accurately identifies mutually exclusive gene sets than existing methods and is at least two orders of magnitude faster than most methods. By applying MEScan to data from four different cancer types and pan-cancer, we have identified several biologically meaningful mutually exclusive gene sets.
Availability and implementation: MEScan is available as an R package at https://github.com/MarkeyBBSRF/MEScan.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures

References
-
- Best S.A. et al. (2018) Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab., 27, 935–943. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
