

Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements
- PMID: 33166999
- PMCID: PMC7736786
- DOI: 10.1093/nar/gkaa1053
Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements
Abstract
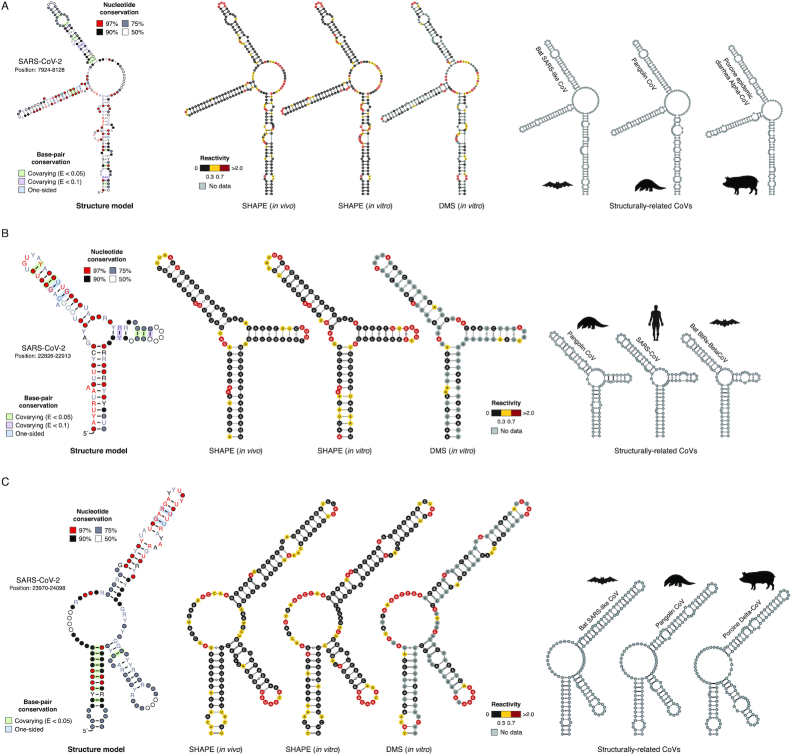
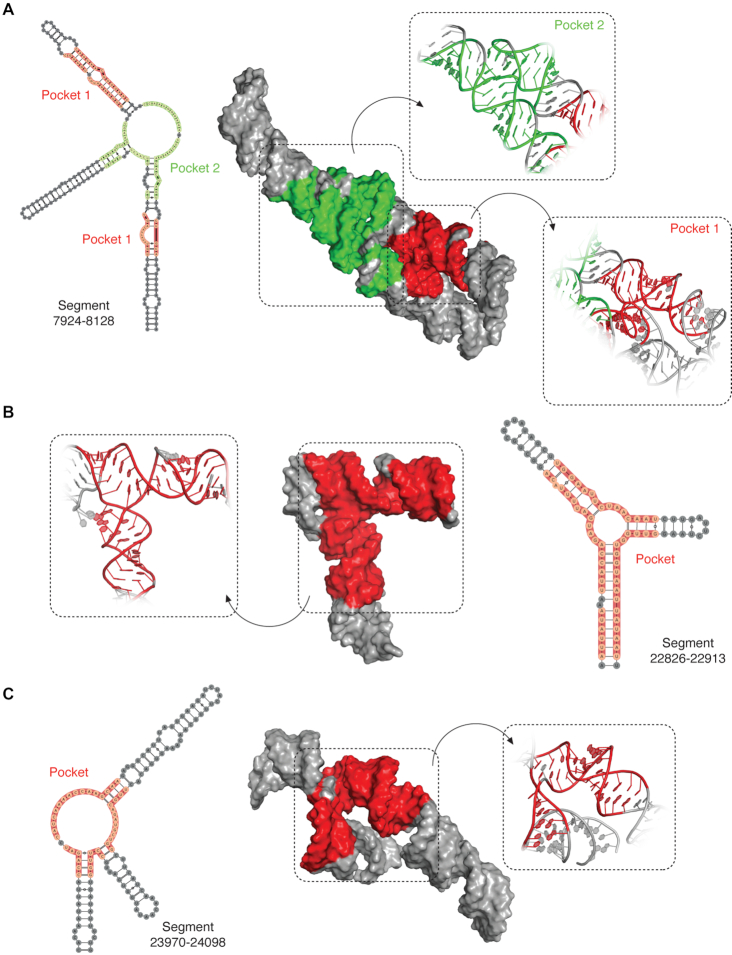
SARS-CoV-2 is a betacoronavirus with a linear single-stranded, positive-sense RNA genome, whose outbreak caused the ongoing COVID-19 pandemic. The ability of coronaviruses to rapidly evolve, adapt, and cross species barriers makes the development of effective and durable therapeutic strategies a challenging and urgent need. As for other RNA viruses, genomic RNA structures are expected to play crucial roles in several steps of the coronavirus replication cycle. Despite this, only a handful of functionally-conserved coronavirus structural RNA elements have been identified to date. Here, we performed RNA structure probing to obtain single-base resolution secondary structure maps of the full SARS-CoV-2 coronavirus genome both in vitro and in living infected cells. Probing data recapitulate the previously described coronavirus RNA elements (5' UTR and s2m), and reveal new structures. Of these, ∼10.2% show significant covariation among SARS-CoV-2 and other coronaviruses, hinting at their functionally-conserved role. Secondary structure-restrained 3D modeling of these segments further allowed for the identification of putative druggable pockets. In addition, we identify a set of single-stranded segments in vivo, showing high sequence conservation, suitable for the development of antisense oligonucleotide therapeutics. Collectively, our work lays the foundation for the development of innovative RNA-targeted therapeutic strategies to fight SARS-related infections.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
