

Comparative assessments of indel annotations in healthy and cancer genomes with next-generation sequencing data
- PMID: 33167946
- PMCID: PMC7653722
- DOI: 10.1186/s12920-020-00818-6
Comparative assessments of indel annotations in healthy and cancer genomes with next-generation sequencing data
Abstract
Background: Insertion and deletion (indel) is one of the major variation types in human genomes. Accurate annotation of indels is of paramount importance in genetic variation analysis and investigation of their roles in human diseases. Previous studies revealed a high number of false positives from existing indel calling methods, which limits downstream analyses of the effects of indels on both healthy and disease genomes. In this study, we evaluated seven commonly used general indel calling programs for germline indels and four somatic indel calling programs through comparative analysis to investigate their common features and differences and to explore ways to improve indel annotation accuracy.
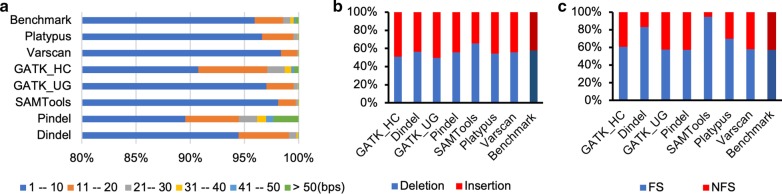
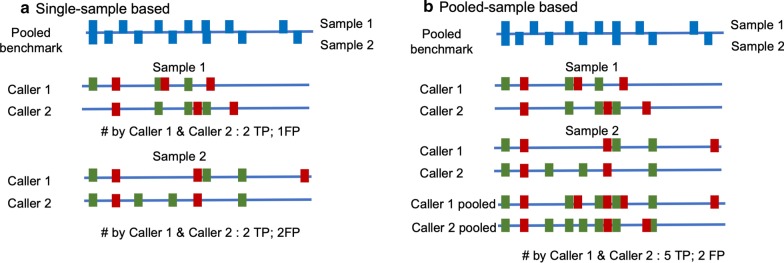
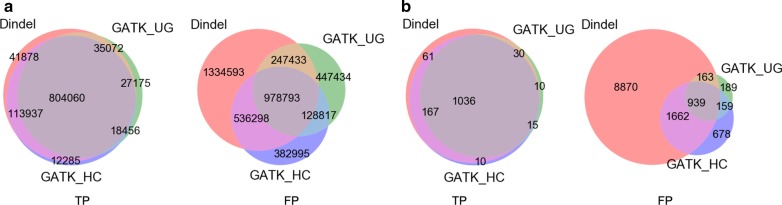
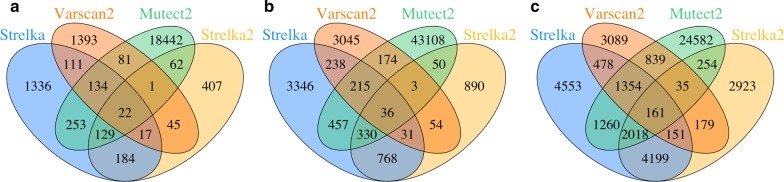
Methods: In our comparative analysis, we adopted a more stringent evaluation approach by considering both the indel positions and the indel types (insertion or deletion sequences) between the samples and the reference set. In addition, we applied an efficient way to use a benchmark for improved performance comparisons for the general indel calling programs RESULTS: We found that germline indels in healthy genomes derived by combining several indel calling tools could help remove a large number of false positive indels from individual programs without compromising the number of true positives. The performance comparisons of somatic indel calling programs are more complicated due to the lack of a reliable and comprehensive benchmark. Nevertheless our results revealed large variations among the programs and among cancer types.
Conclusions: While more accurate indel calling programs are needed, we found that the performance for germline indel annotations can be improved by combining the results from several programs. In addition, well-designed benchmarks for both germline and somatic indels are key in program development and evaluations.
Keywords: Cancer; Deletion; Germline variants; Indel; Insertion; Somatic variants.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–745. doi: 10.1093/nar/gkv1189. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
