

PINK1/PARKIN signalling in neurodegeneration and neuroinflammation
- PMID: 33168089
- PMCID: PMC7654589
- DOI: 10.1186/s40478-020-01062-w
PINK1/PARKIN signalling in neurodegeneration and neuroinflammation
Abstract
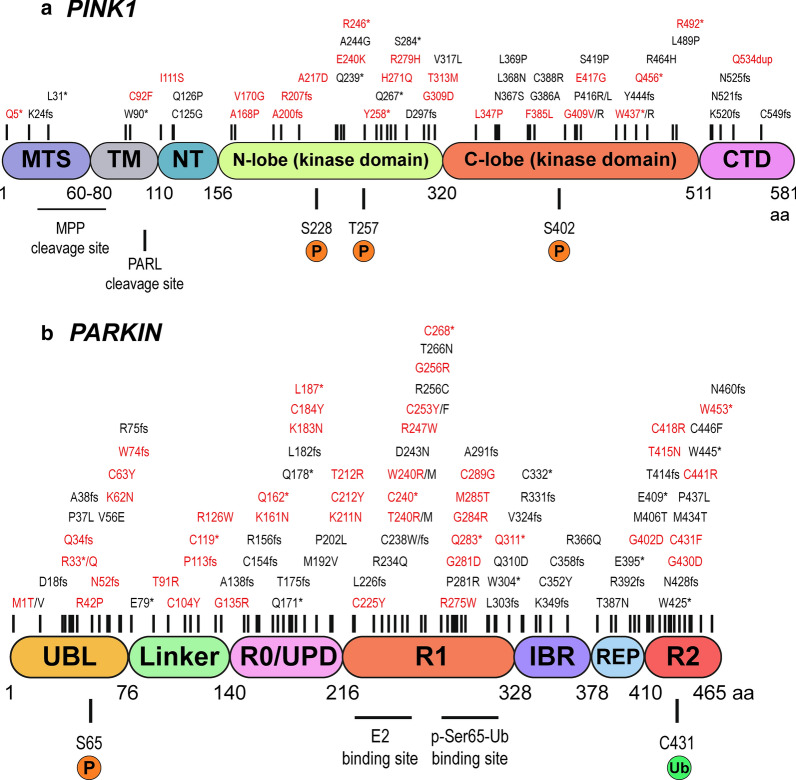
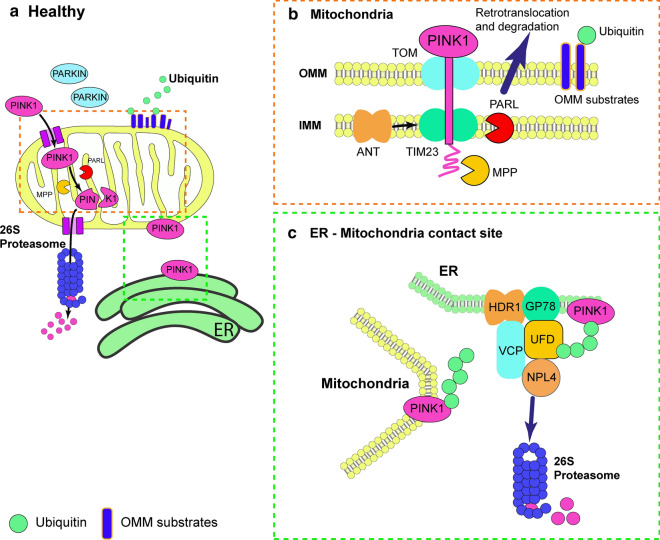
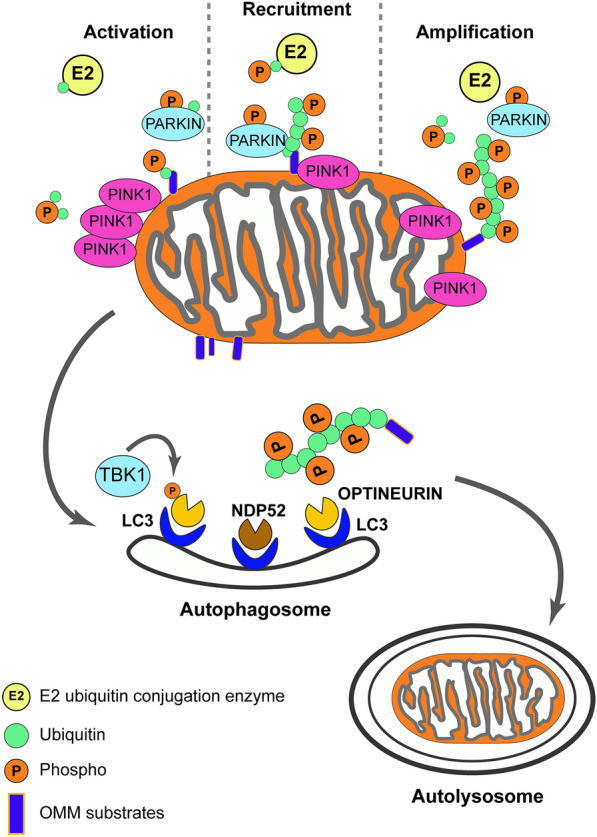
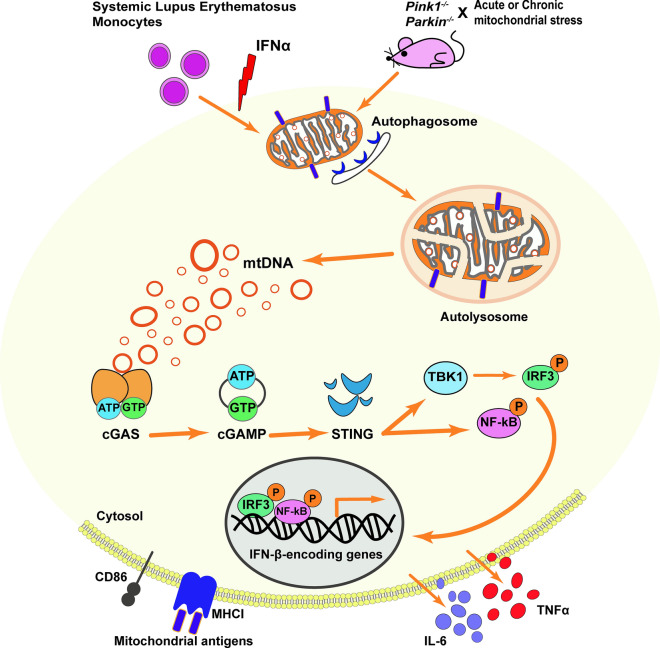
Mutations in the PTEN-induced kinase 1 (PINK1) and Parkin RBR E3 ubiquitin-protein ligase (PARKIN) genes are associated with familial forms of Parkinson's disease (PD). PINK1, a protein kinase, and PARKIN, an E3 ubiquitin ligase, control the specific elimination of dysfunctional or superfluous mitochondria, thus fine-tuning mitochondrial network and preserving energy metabolism. PINK1 regulates PARKIN translocation in impaired mitochondria and drives their removal via selective autophagy, a process known as mitophagy. As knowledge obtained using different PINK1 and PARKIN transgenic animal models is being gathered, growing evidence supports the contribution of mitophagy impairment to several human pathologies, including PD and Alzheimer's diseases (AD). Therefore, therapeutic interventions aiming to modulate PINK1/PARKIN signalling might have the potential to treat these diseases. In this review, we will start by discussing how the interplay of PINK1 and PARKIN signalling helps mediate mitochondrial physiology. We will continue by debating the role of mitochondrial dysfunction in disorders such as amyotrophic lateral sclerosis, Alzheimer's, Huntington's and Parkinson's diseases, as well as eye diseases such as age-related macular degeneration and glaucoma, and the causative factors leading to PINK1/PARKIN-mediated neurodegeneration and neuroinflammation. Finally, we will discuss PINK1/PARKIN gene augmentation possibilities with a particular focus on AD, PD and glaucoma.
Keywords: Alzheimer’s disease; Mitophagy; Neurodegeneration; PARKIN; PINK1; Parkinson’s disease.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Benda C. Ueber dier Spermatogenese de Verbebraten und höherer Evertebraten, II. Theil: die Histiogenese der Spermien. Arch Anat Physiol Arch Anat Physiol. 1898;73:393–398.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
