

Uncovering cancer gene regulation by accurate regulatory network inference from uninformative data
- PMID: 33168813
- PMCID: PMC7652823
- DOI: 10.1038/s41540-020-00154-6
Uncovering cancer gene regulation by accurate regulatory network inference from uninformative data
Abstract
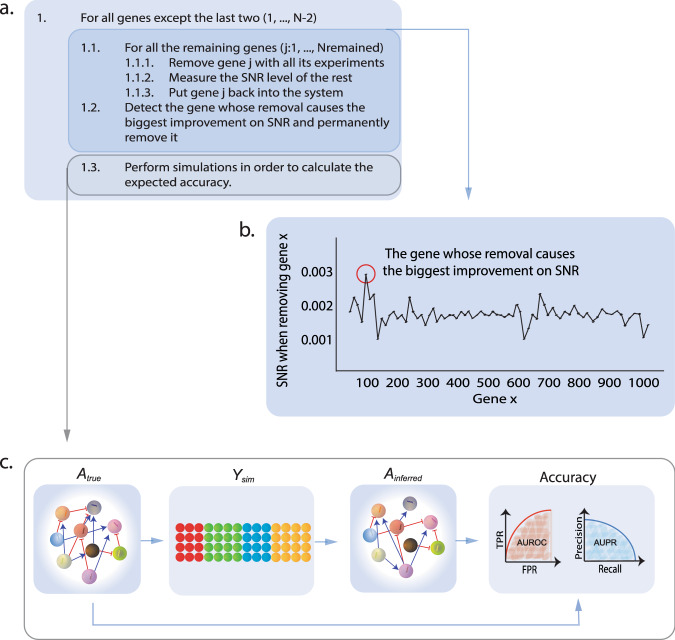
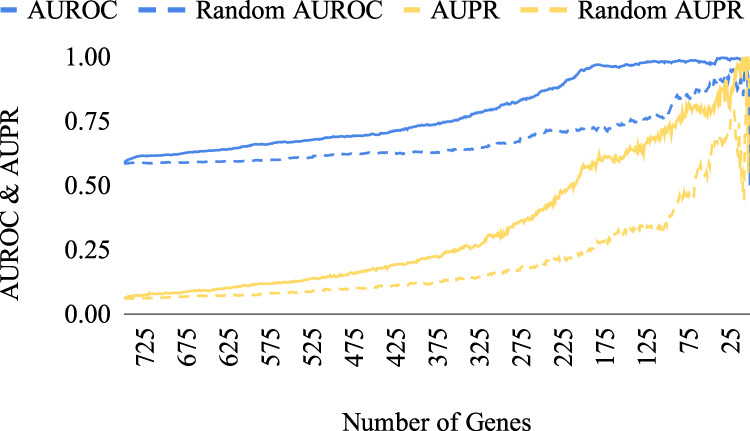
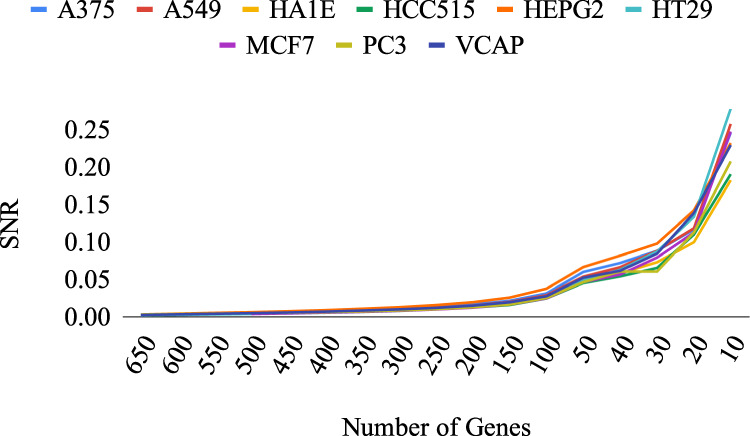
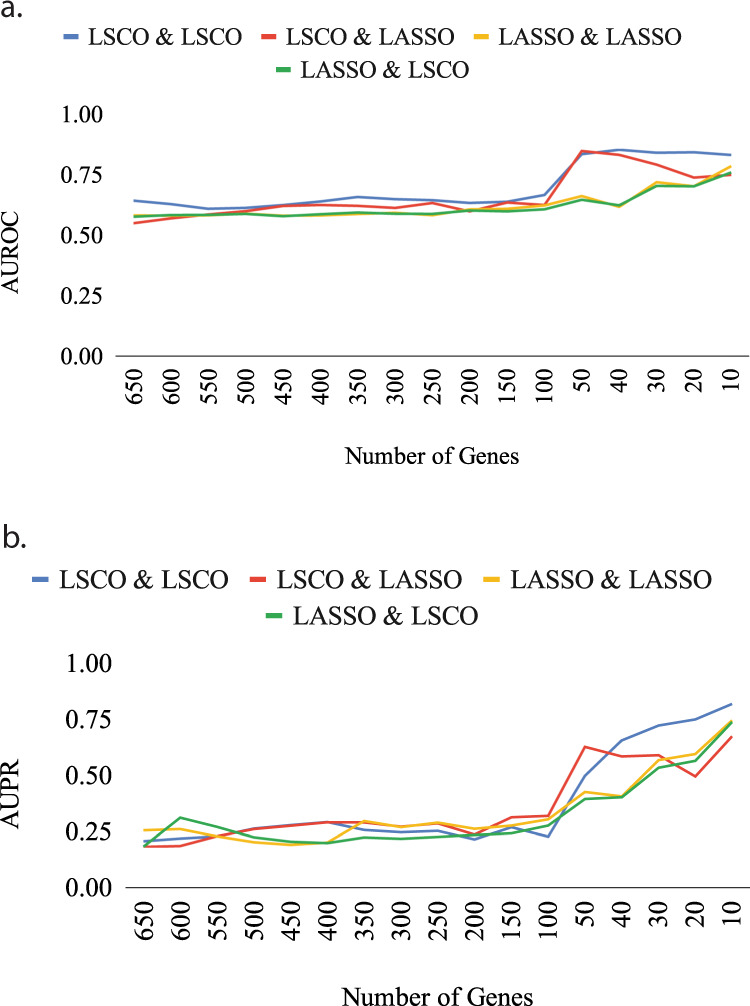
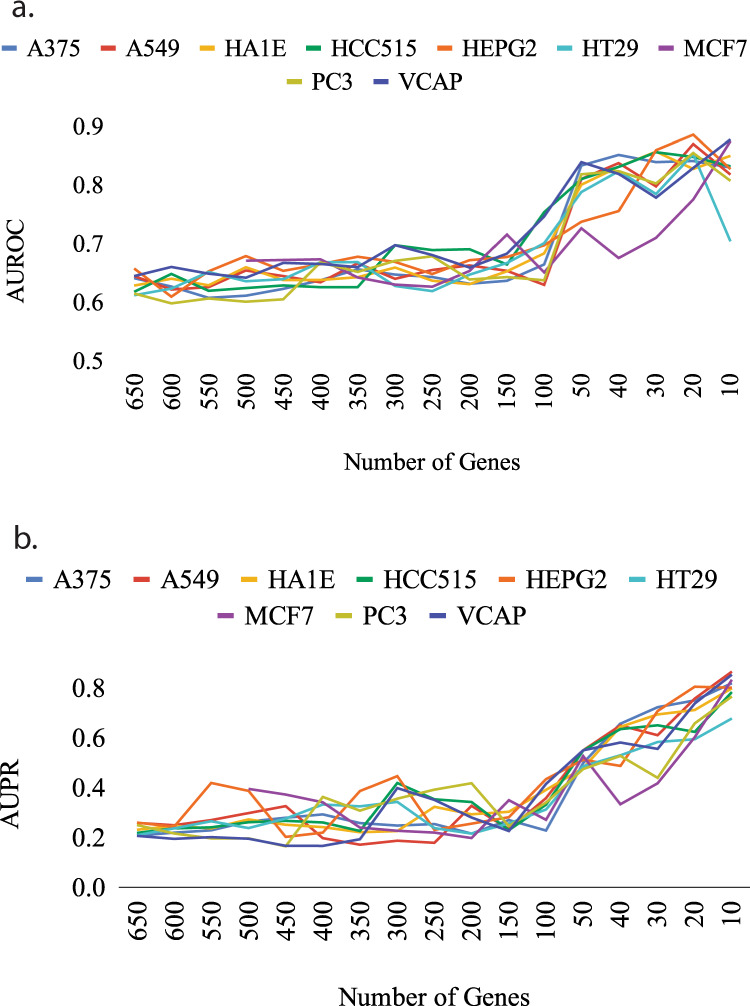
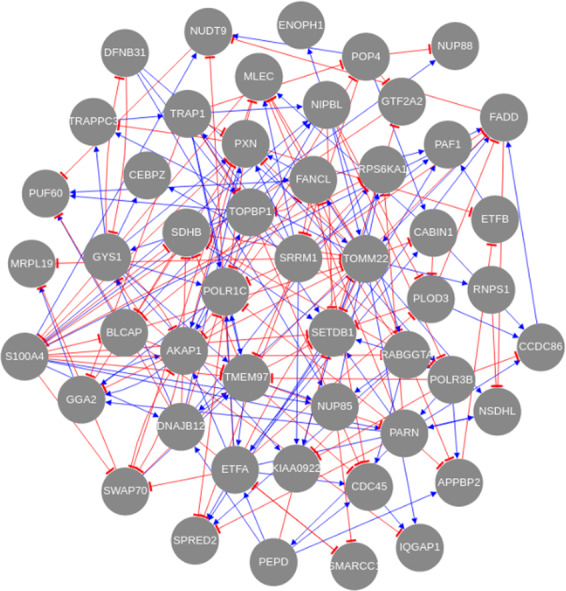
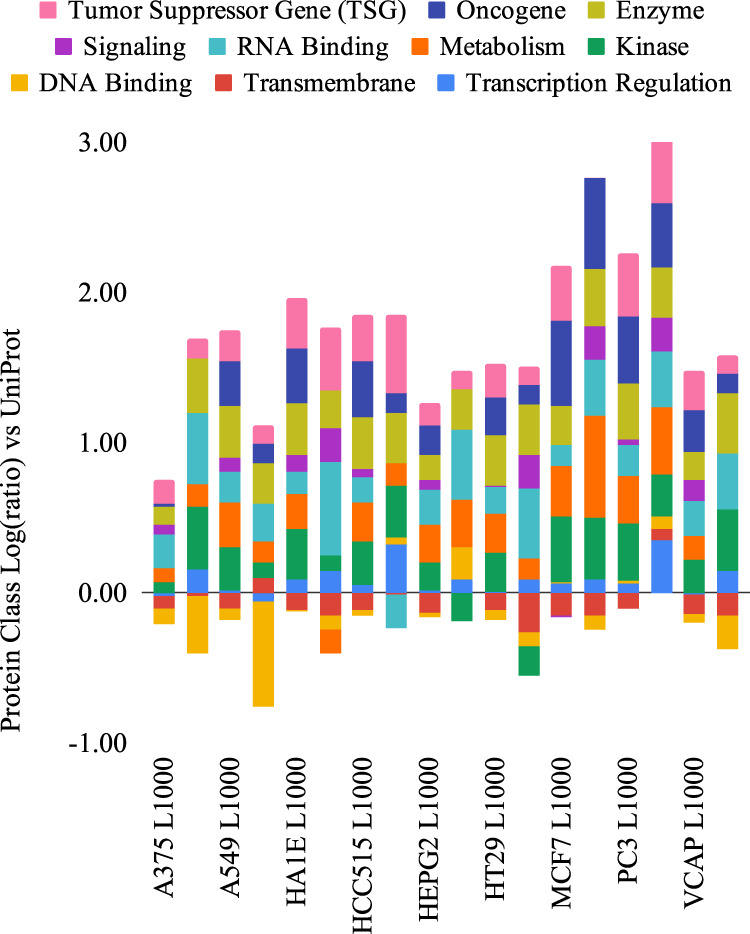
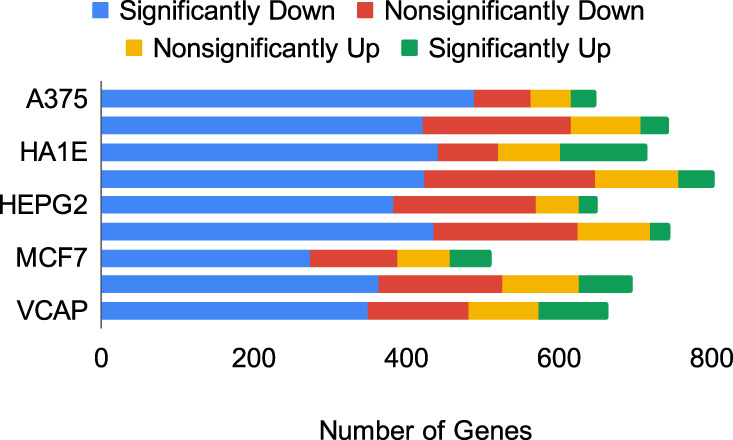
The interactions among the components of a living cell that constitute the gene regulatory network (GRN) can be inferred from perturbation-based gene expression data. Such networks are useful for providing mechanistic insights of a biological system. In order to explore the feasibility and quality of GRN inference at a large scale, we used the L1000 data where ~1000 genes have been perturbed and their expression levels have been quantified in 9 cancer cell lines. We found that these datasets have a very low signal-to-noise ratio (SNR) level causing them to be too uninformative to infer accurate GRNs. We developed a gene reduction pipeline in which we eliminate uninformative genes from the system using a selection criterion based on SNR, until reaching an informative subset. The results show that our pipeline can identify an informative subset in an overall uninformative dataset, allowing inference of accurate subset GRNs. The accurate GRNs were functionally characterized and potential novel cancer-related regulatory interactions were identified.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996;58:267–288.
-
- Nordling, T. E. M. Robust inference of gene regulatory networks, PhD thesis, KTH School of Electrical Engineering, Automatic Control Lab (2013).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
