

Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease
- PMID: 33168999
- PMCID: PMC9782736
- DOI: 10.1038/s41436-020-01026-4
Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease
Abstract
Purpose: To investigate the prevalence of biallelic PKD1 and PKD2 variants underlying very early onset (VEO) polycystic kidney disease (PKD) in a large international pediatric cohort referred for clinical indications over a 10-year period (2010-2020).
Methods: All samples were tested by Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA) of PKD1 and PKD2 genes and/or a next-generation sequencing panel of 15 additional cystic genes including PKHD1 and HNF1B. Two patients underwent exome or genome sequencing.
Results: Likely causative PKD1 or PKD2 variants were detected in 30 infants with PKD-VEO, 16 of whom presented in utero. Twenty-one of 30 (70%) had two variants with biallelic in trans inheritance confirmed in 16/21, 1 infant had biallelic PKD2 variants, and 2 infants had digenic PKD1/PKD2 variants. There was no known family history of ADPKD in 13 families (43%) and a de novo pathogenic variant was confirmed in 6 families (23%).
Conclusion: We report a high prevalence of hypomorphic PKD1 variants and likely biallelic disease in infants presenting with PKD-VEO with major implications for reproductive counseling. The diagnostic interpretation and reporting of these variants however remains challenging using current American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) and Association of Clinical Genetic Science (ACGS) variant classification guidelines in PKD-VEO and other diseases affected by similar variants with incomplete penetrance.
Conflict of interest statement
COMPETING INTERESTS
The authors declare no conflicts of interest.
Figures

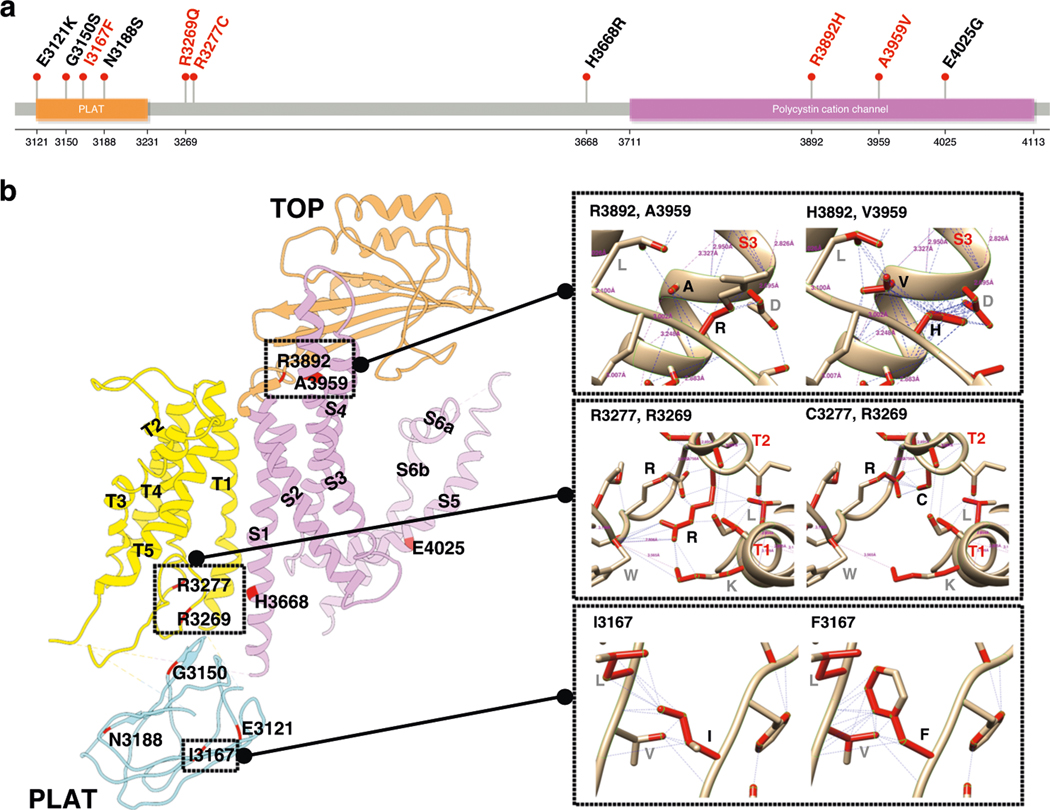
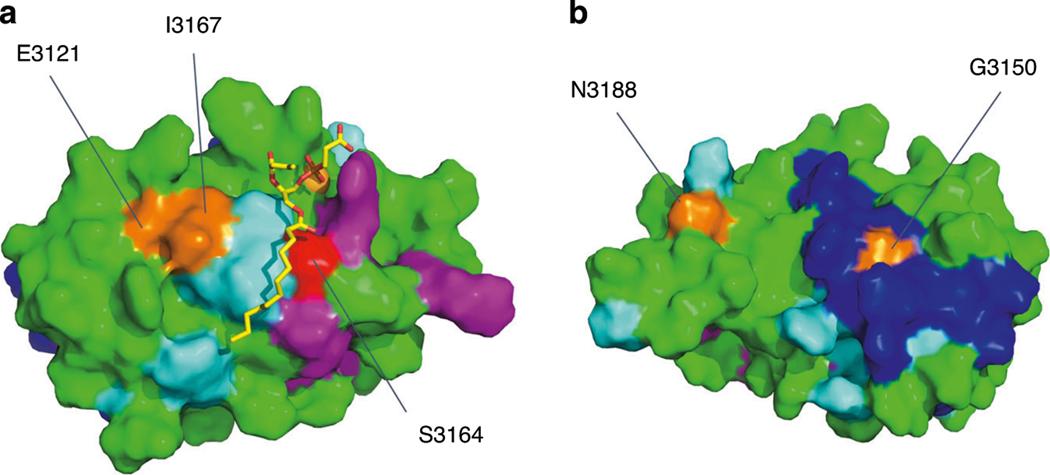
References
-
- Ong AC, Devuyst O, Knebelmann B, Walz G, ERA-EDTA Working Group for Inherited Kidney Diseases. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385:1993–2002. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
