

16p11.2 microdeletion imparts transcriptional alterations in human iPSC-derived models of early neural development
- PMID: 33169669
- PMCID: PMC7695459
- DOI: 10.7554/eLife.58178
16p11.2 microdeletion imparts transcriptional alterations in human iPSC-derived models of early neural development
Abstract
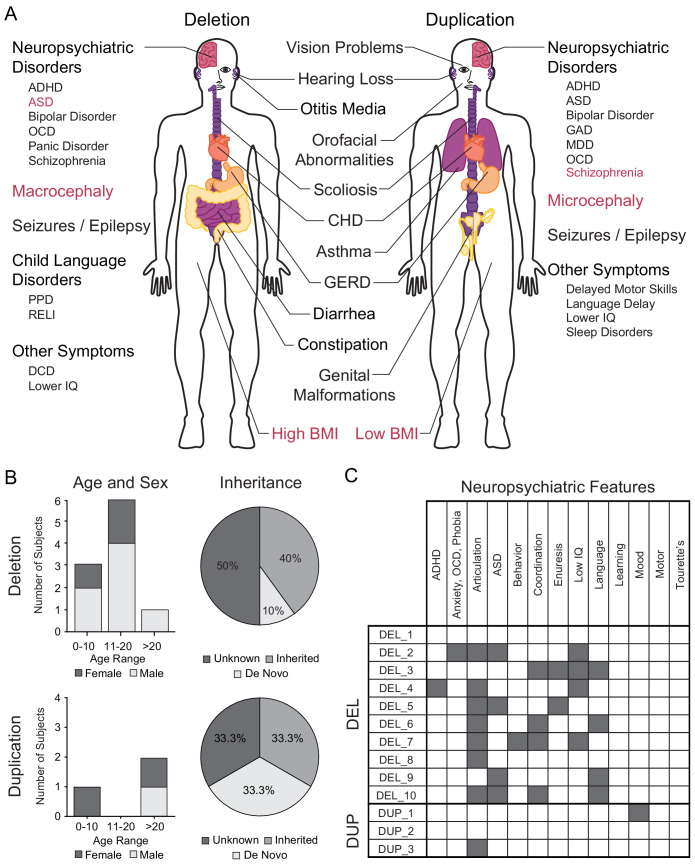
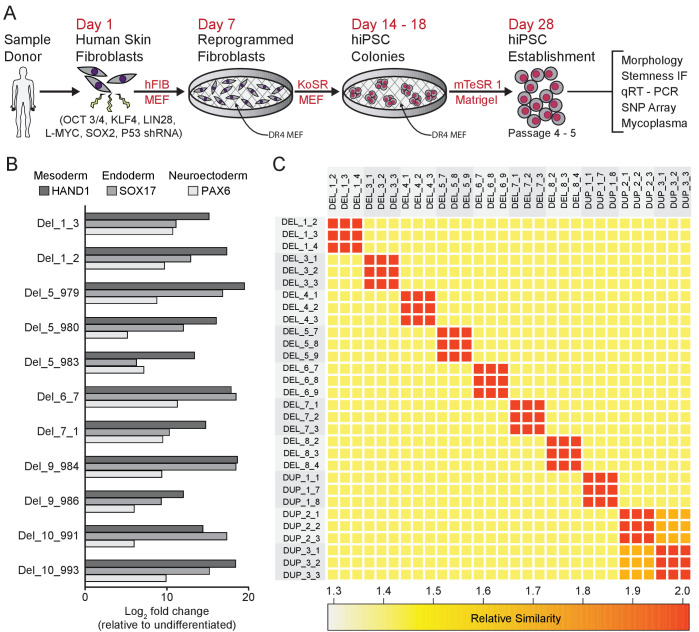
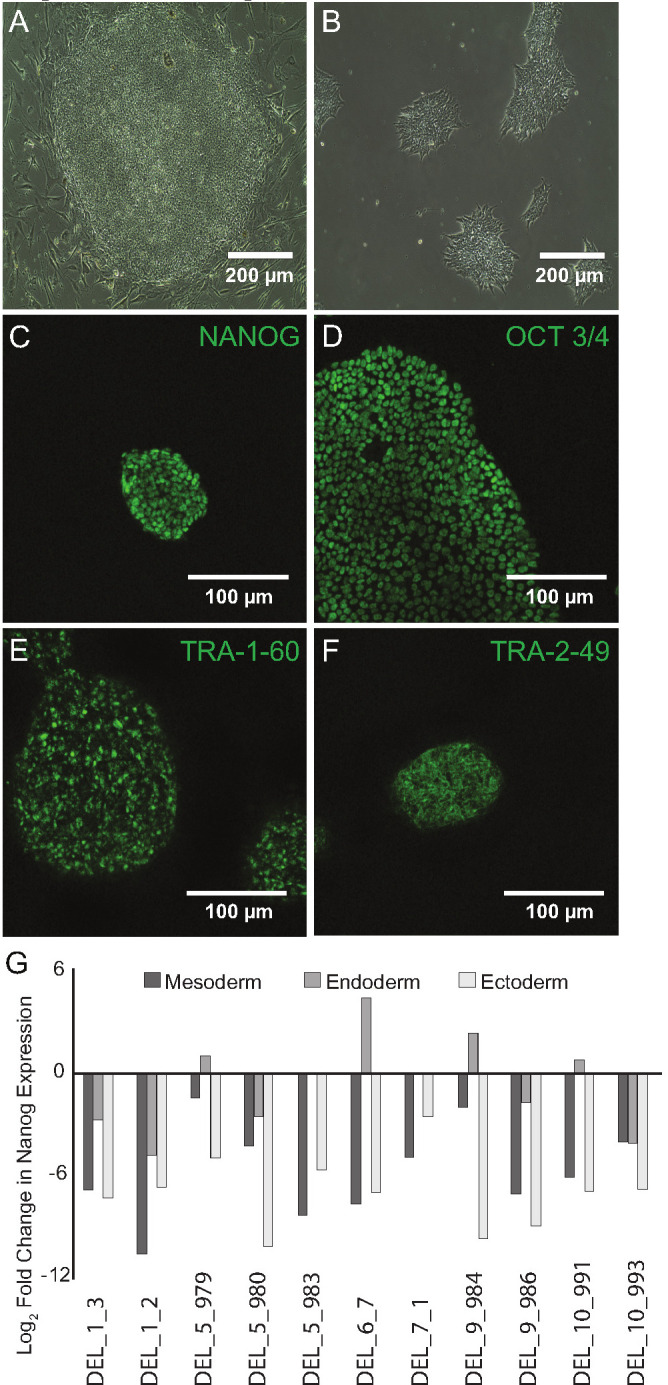
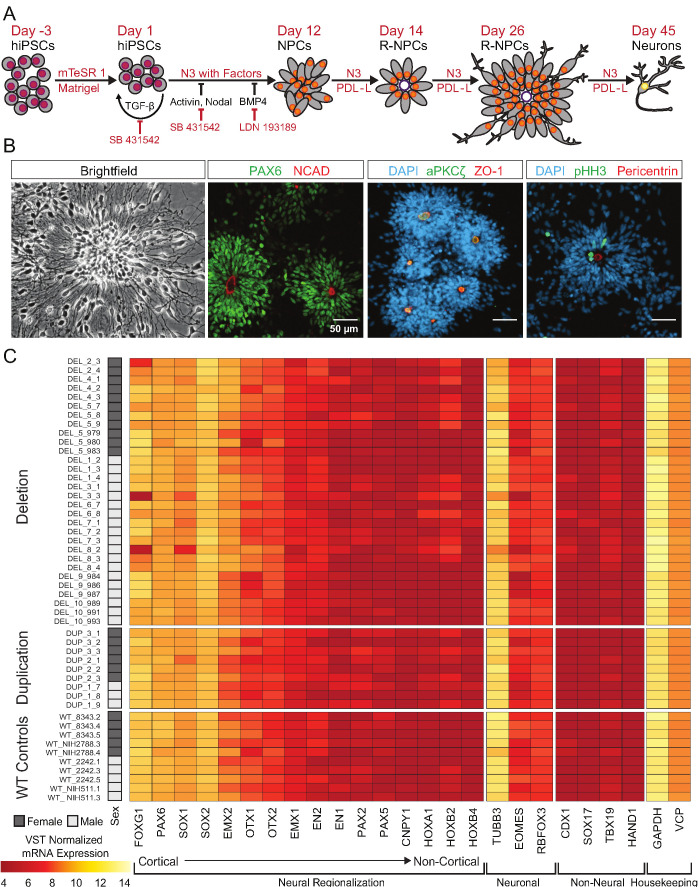
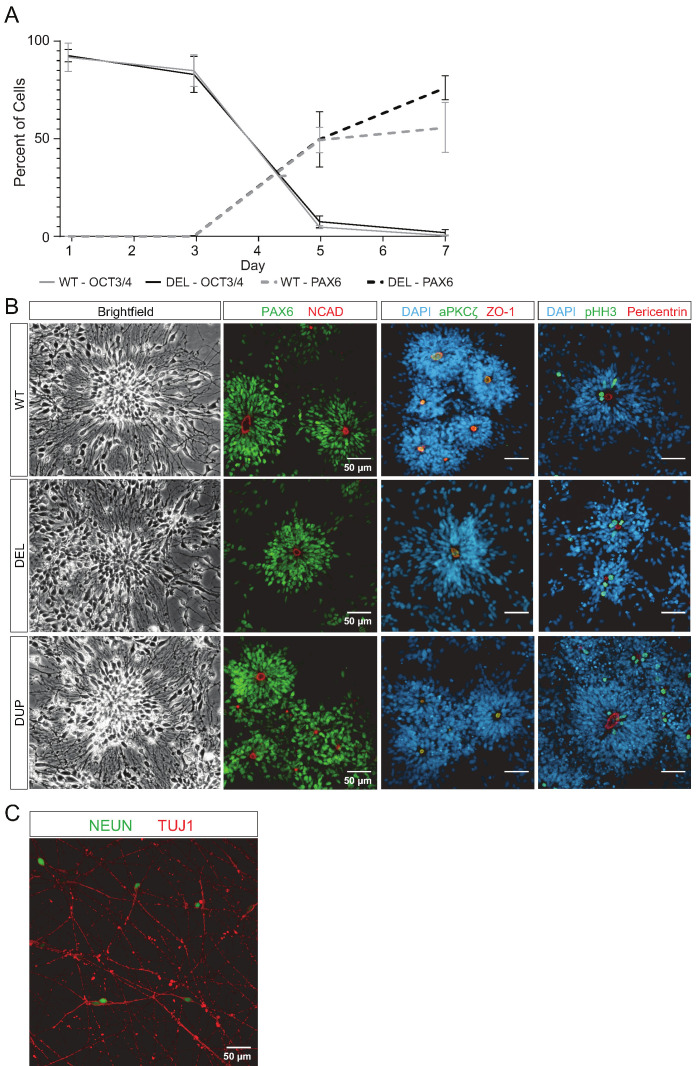
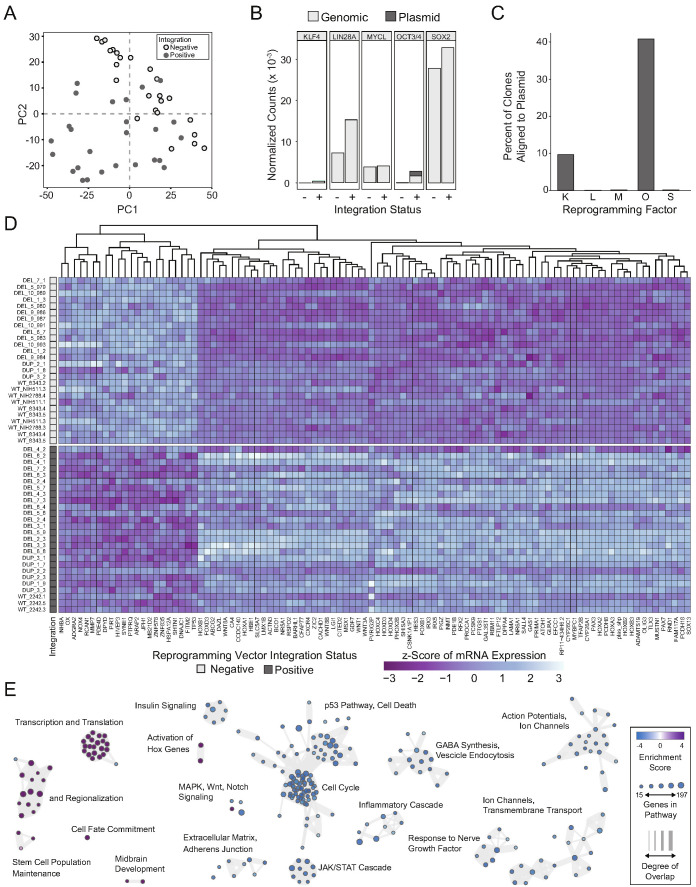
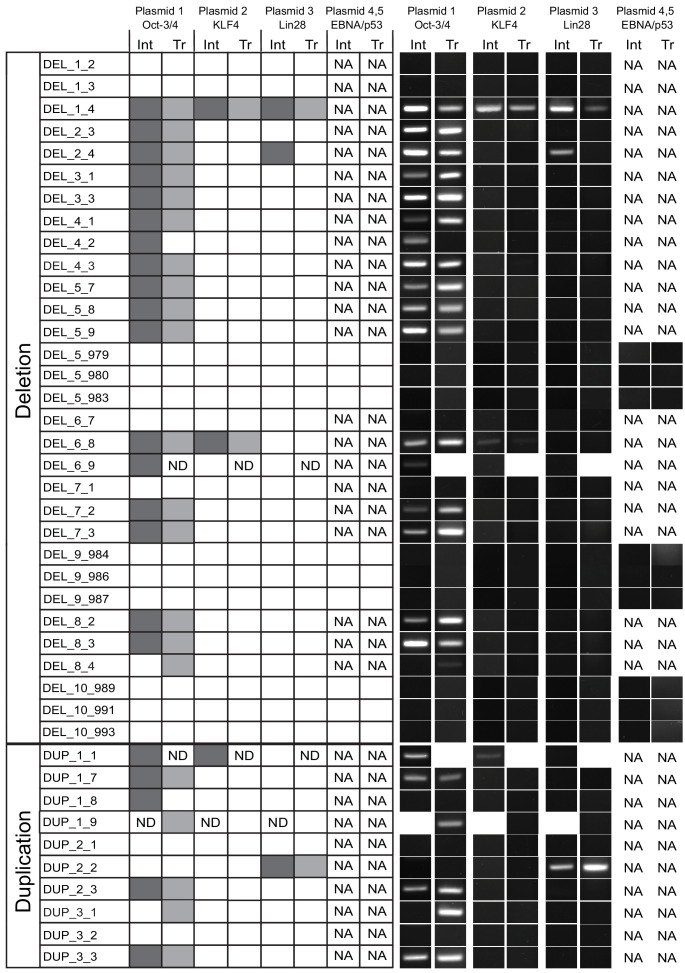
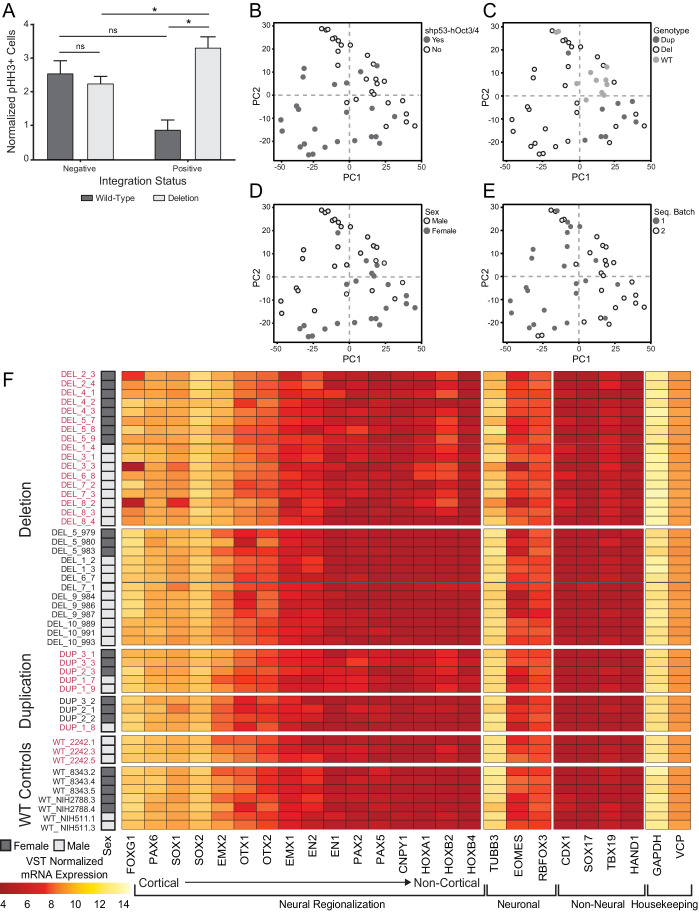
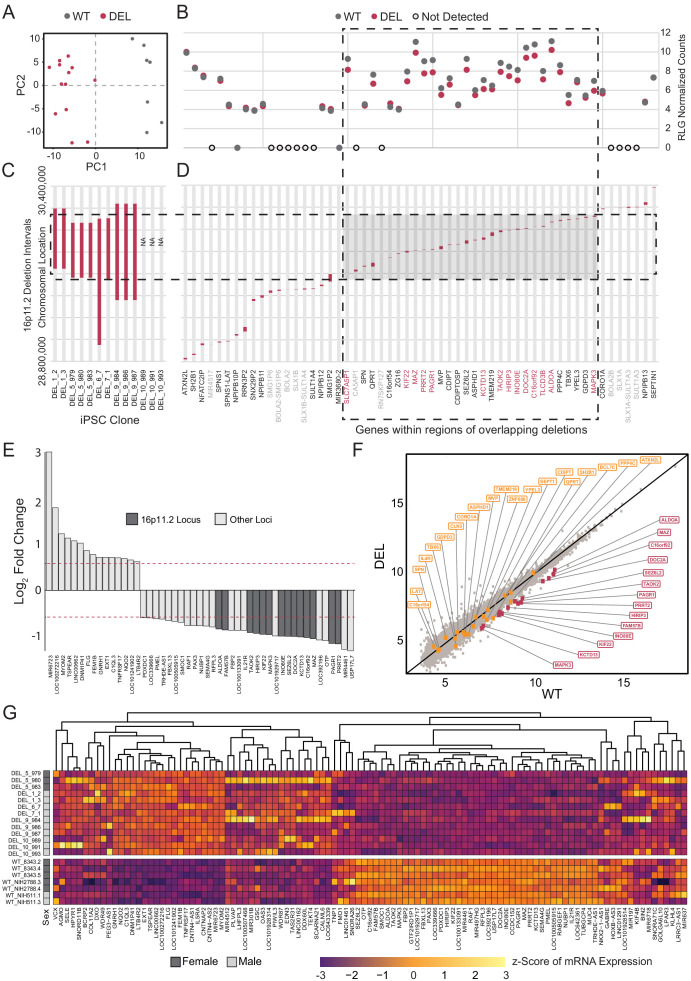
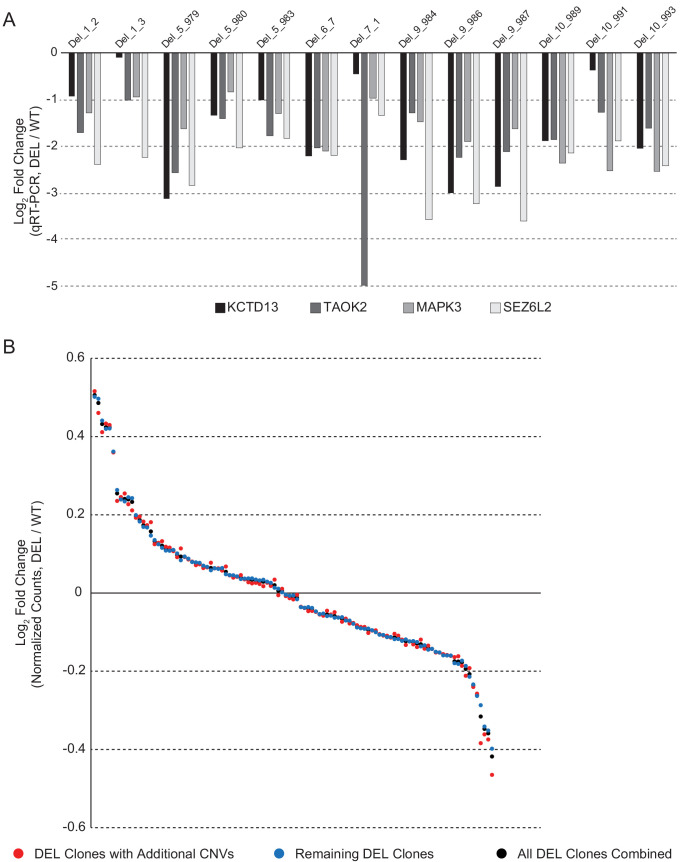
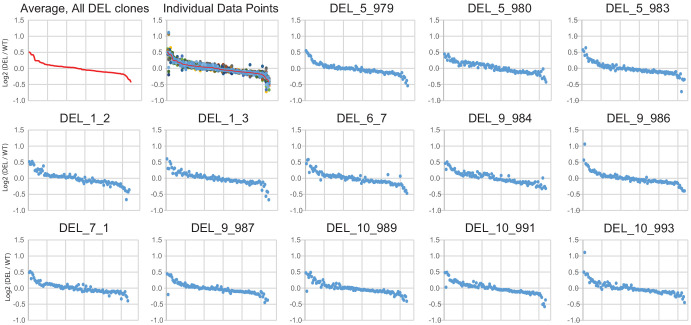
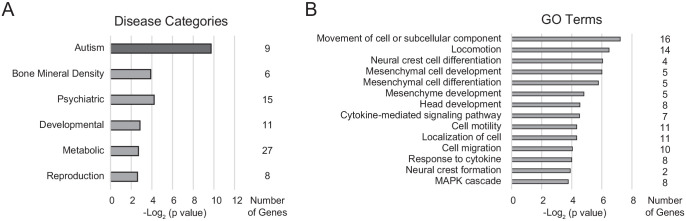
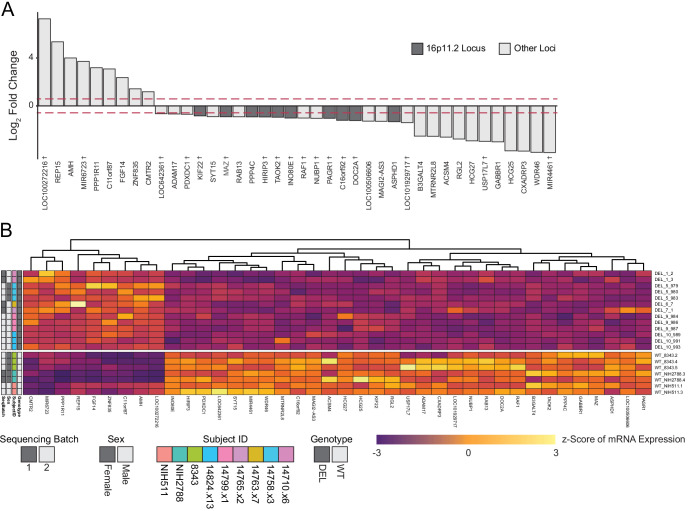
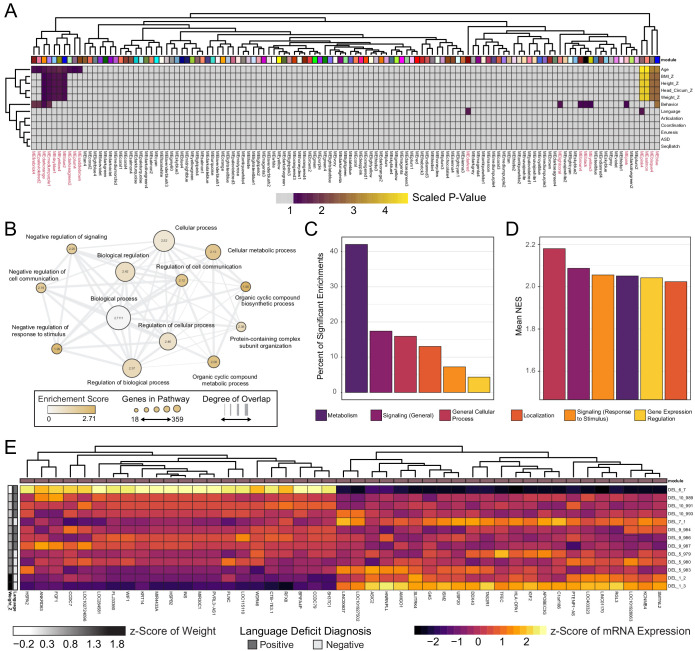
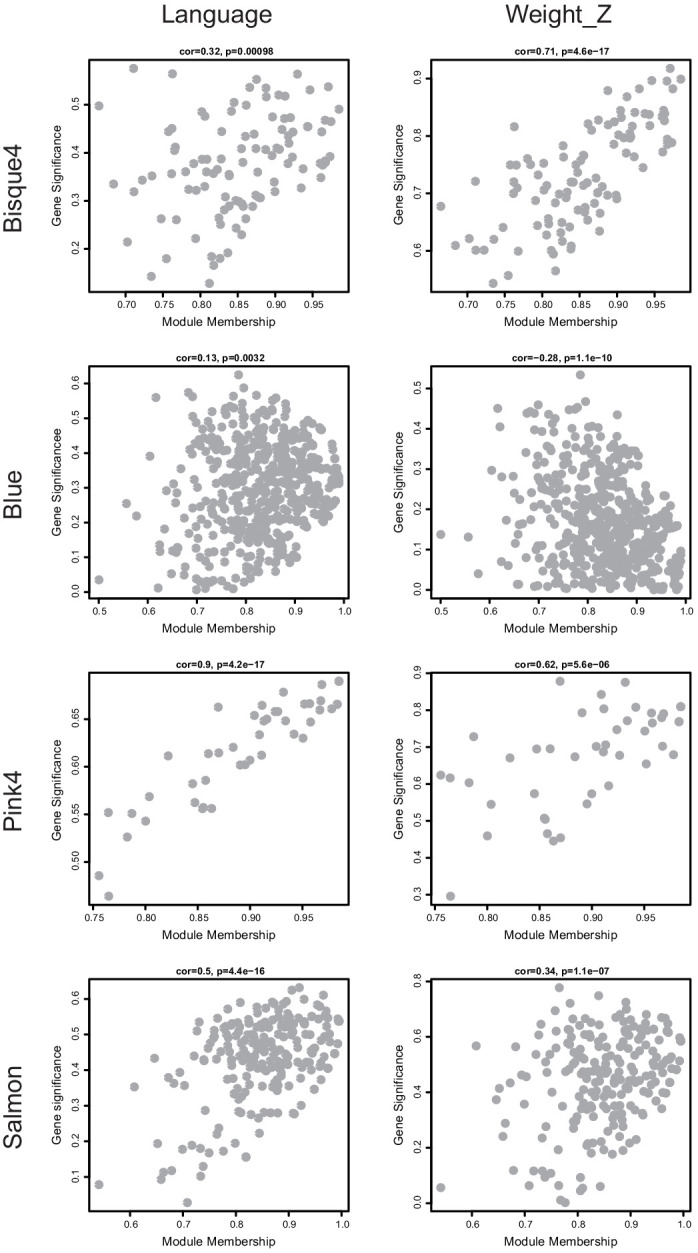
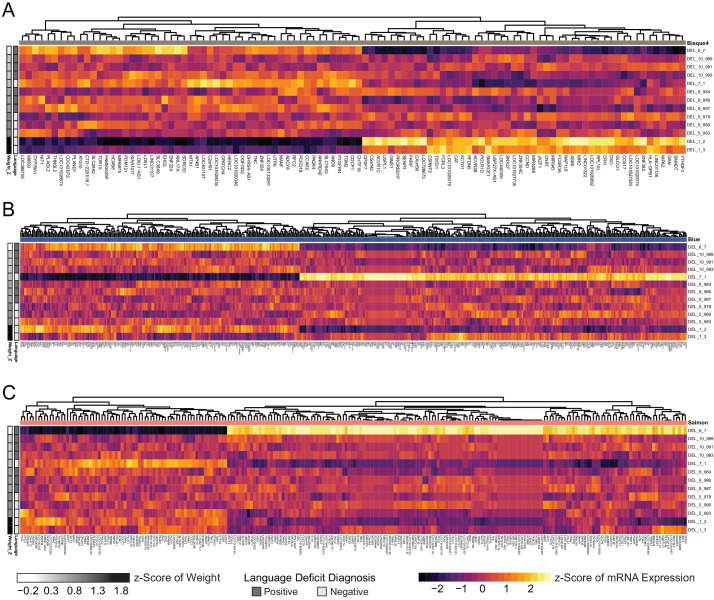
Microdeletions and microduplications of the 16p11.2 chromosomal locus are associated with syndromic neurodevelopmental disorders and reciprocal physiological conditions such as macro/microcephaly and high/low body mass index. To facilitate cellular and molecular investigations into these phenotypes, 65 clones of human induced pluripotent stem cells (hiPSCs) were generated from 13 individuals with 16p11.2 copy number variations (CNVs). To ensure these cell lines were suitable for downstream mechanistic investigations, a customizable bioinformatic strategy for the detection of random integration and expression of reprogramming vectors was developed and leveraged towards identifying a subset of 'footprint'-free hiPSC clones. Transcriptomic profiling of cortical neural progenitor cells derived from these hiPSCs identified alterations in gene expression patterns which precede morphological abnormalities reported at later neurodevelopmental stages. Interpreting clinical information-available with the cell lines by request from the Simons Foundation Autism Research Initiative-with this transcriptional data revealed disruptions in gene programs related to both nervous system function and cellular metabolism. As demonstrated by these analyses, this publicly available resource has the potential to serve as a powerful medium for probing the etiology of developmental disorders associated with 16p11.2 CNVs.
Keywords: 16p11.2; copy number variation; corticogenesis; human; iPSC; neurodevelopment; neuroscience; regenerative medicine; stem cells.
© 2020, Roth et al.
Conflict of interest statement
JR, KM, AA, VM, HG, YV, SW, CC, JF, KL, RD, TP No competing interests declared
Figures

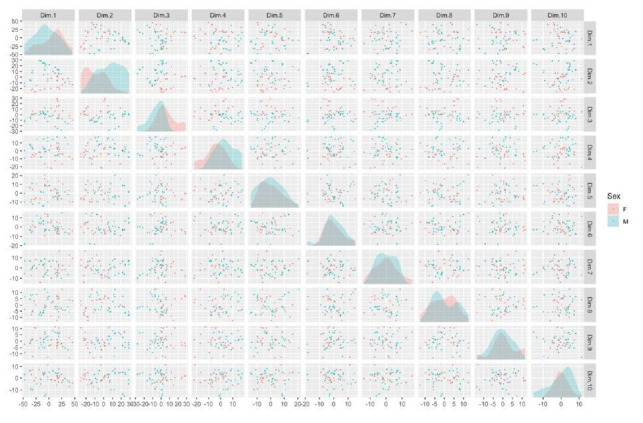
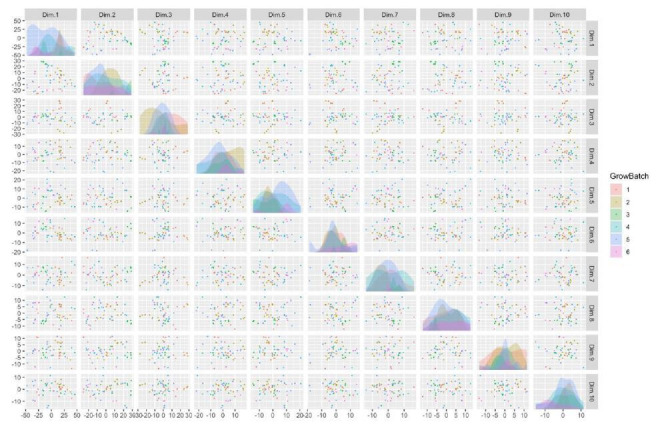
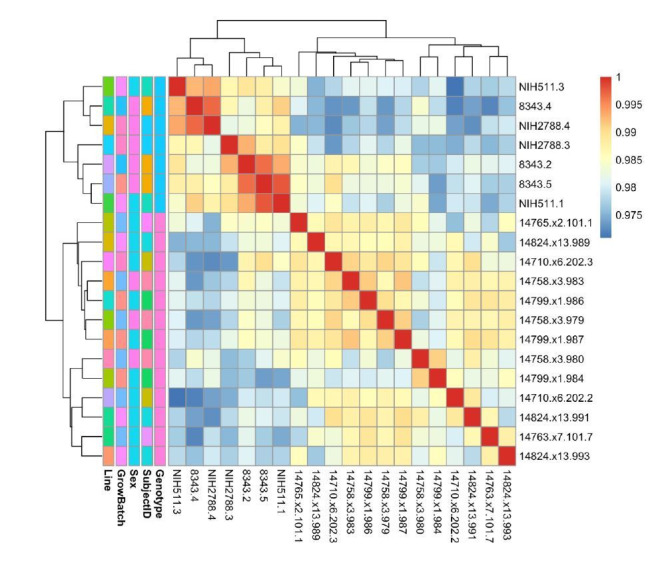
Similar articles
-
Characterizing Sleep Problems in 16p11.2 Deletion and Duplication.J Autism Dev Disord. 2023 Apr;53(4):1462-1475. doi: 10.1007/s10803-021-05311-2. Epub 2021 Oct 11. J Autism Dev Disord. 2023. PMID: 34633643
-
Clinical and Genetic Study of Three Inherited Microdeletions of Chromosome 16p11.2.Altern Ther Health Med. 2024 Mar;30(3):190-192. Altern Ther Health Med. 2024. PMID: 37856819
-
Tissue- and cell-type-specific molecular and functional signatures of 16p11.2 reciprocal genomic disorder across mouse brain and human neuronal models.Am J Hum Genet. 2022 Oct 6;109(10):1789-1813. doi: 10.1016/j.ajhg.2022.08.012. Epub 2022 Sep 23. Am J Hum Genet. 2022. PMID: 36152629 Free PMC article.
-
16p11.2 Copy Number Variations and Neurodevelopmental Disorders.Trends Neurosci. 2020 Nov;43(11):886-901. doi: 10.1016/j.tins.2020.09.001. Epub 2020 Sep 28. Trends Neurosci. 2020. PMID: 32993859 Free PMC article. Review.
-
A Hypothesis: Metabolic Contributions to 16p11.2 Deletion Syndrome.Bioessays. 2025 Mar;47(3):e202400177. doi: 10.1002/bies.202400177. Epub 2024 Dec 29. Bioessays. 2025. PMID: 39988938 Free PMC article. Review.
Cited by
-
oFlowSeq: a quantitative approach to identify protein coding mutations affecting cell type enrichment using mosaic CRISPR-Cas9 edited cerebral organoids.Hum Genet. 2023 Aug;142(8):1281-1291. doi: 10.1007/s00439-023-02534-4. Epub 2023 Mar 6. Hum Genet. 2023. PMID: 36877372 Free PMC article.
-
Custom-engineered hydrogels for delivery of human iPSC-derived neurons into the injured cervical spinal cord.Biomaterials. 2024 Mar;305:122400. doi: 10.1016/j.biomaterials.2023.122400. Epub 2023 Nov 17. Biomaterials. 2024. PMID: 38134472 Free PMC article.
-
Spatiotemporal 22q11.21 Protein Network Implicates DGCR8-Dependent MicroRNA Biogenesis as a Risk for Late-Fetal Cortical Development in Psychiatric Diseases.Life (Basel). 2021 May 31;11(6):514. doi: 10.3390/life11060514. Life (Basel). 2021. PMID: 34073122 Free PMC article.
-
Identifying foetal forebrain interneurons as a target for monogenic autism risk factors and the polygenic 16p11.2 microdeletion.BMC Neurosci. 2023 Jan 19;24(1):5. doi: 10.1186/s12868-022-00771-3. BMC Neurosci. 2023. PMID: 36658491 Free PMC article.
-
A resource of induced pluripotent stem cell (iPSC) lines including clinical, genomic, and cellular data from genetically isolated families with mood and psychotic disorders.Transl Psychiatry. 2023 Dec 16;13(1):397. doi: 10.1038/s41398-023-02641-w. Transl Psychiatry. 2023. PMID: 38104115 Free PMC article.
References
-
- Ang LT, Tan AKY, Autio MI, Goh SH, Choo SH, Lee KL, Tan J, Pan B, Lee JJH, Lum JJ, Lim CYY, Yeo IKX, Wong CJY, Liu M, Oh JLL, Chia CPL, Loh CH, Chen A, Chen Q, Weissman IL, Loh KM, Lim B. A roadmap for human liver differentiation from pluripotent stem cells. Cell Reports. 2018;22:2190–2205. doi: 10.1016/j.celrep.2018.01.087. - DOI - PMC - PubMed
-
- Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, Manavalan P, Zhou JQ, Wheeler VC, Levin JZ, Ernst C, Roeder K, Devlin B, Gusella JF, Talkowski ME. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. The American Journal of Human Genetics. 2014;94:870–883. doi: 10.1016/j.ajhg.2014.05.004. - DOI - PMC - PubMed
-
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
