

Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19
- PMID: 33171100
- PMCID: PMC7598382
- DOI: 10.1016/j.cell.2020.10.037
Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19
Abstract
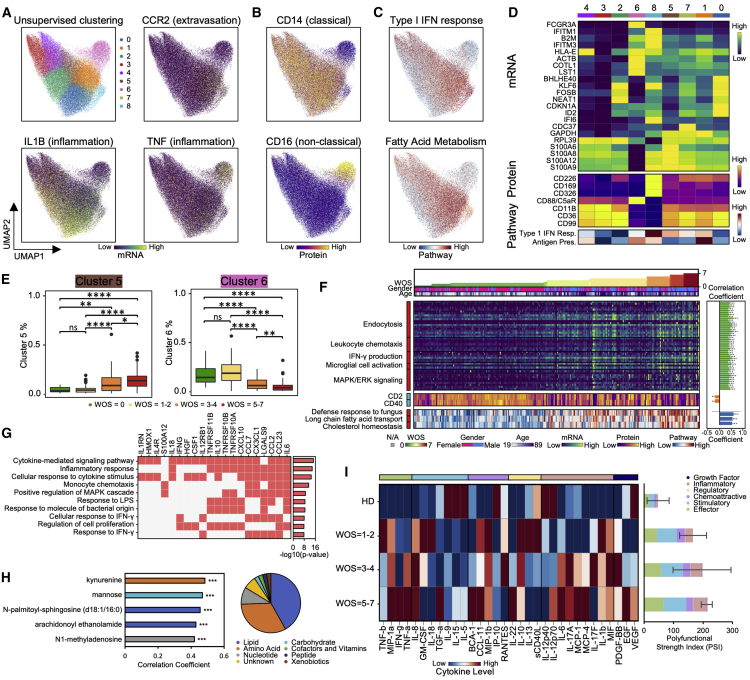
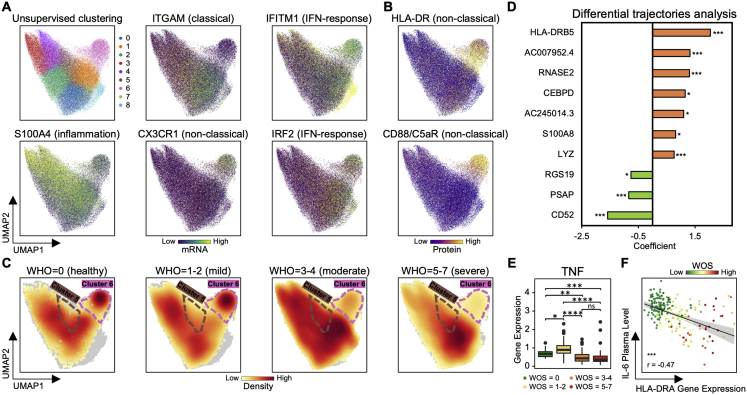
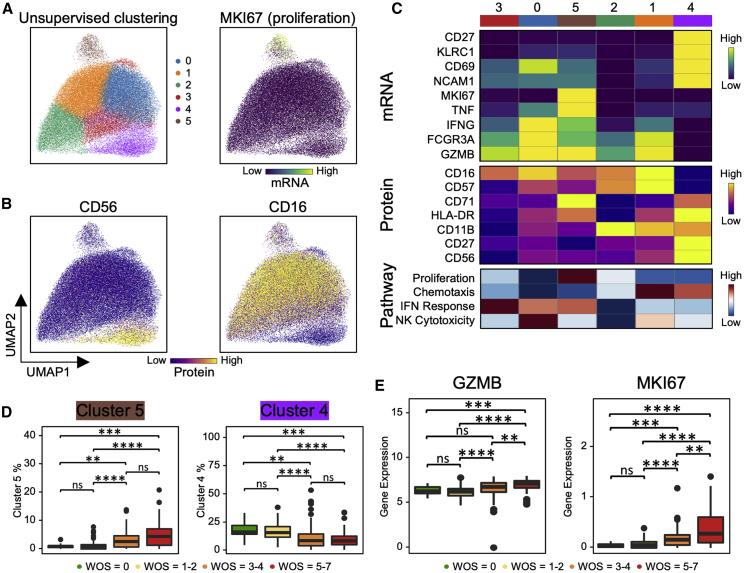
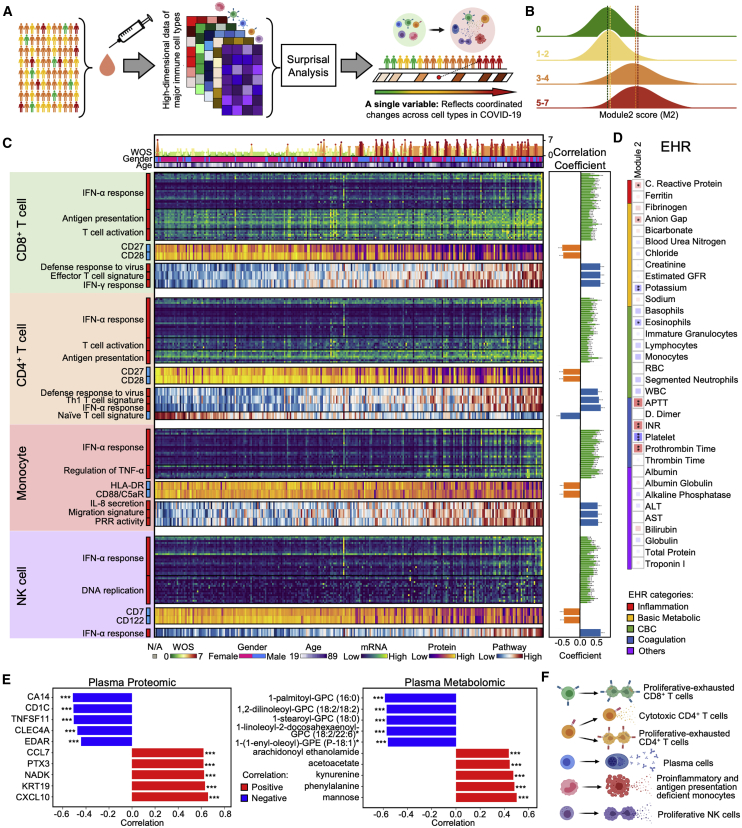
We present an integrated analysis of the clinical measurements, immune cells, and plasma multi-omics of 139 COVID-19 patients representing all levels of disease severity, from serial blood draws collected during the first week of infection following diagnosis. We identify a major shift between mild and moderate disease, at which point elevated inflammatory signaling is accompanied by the loss of specific classes of metabolites and metabolic processes. Within this stressed plasma environment at moderate disease, multiple unusual immune cell phenotypes emerge and amplify with increasing disease severity. We condensed over 120,000 immune features into a single axis to capture how different immune cell classes coordinate in response to SARS-CoV-2. This immune-response axis independently aligns with the major plasma composition changes, with clinical metrics of blood clotting, and with the sharp transition between mild and moderate disease. This study suggests that moderate disease may provide the most effective setting for therapeutic intervention.
Keywords: CITE-seq; COVID-19; immune response; infection; metabolomics; multi-omics; proteomics; single-cell RNA-seq; single-cell TCR-seq; single-cell secretome.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests J.R.H. is founder and board member of Isoplexis and PACT Pharma. M.M.D. is a member of the Scientific Advisory Board of PACT Pharma. J.A.B. is a member of the Scientific Advisory Boards of Arcus, Celsius, and VIR. J.A.B. is a member of the Board of Directors of Rheos and Provention. J.A.B. has recently joined Sonoma Biotherapeutics as President and CEO. Sonoma Biotherapeutics is involved in developing novel Treg-based cell therapies for the treatment of autoimmune diseases. R.G. has received consulting income from Juno Therapeutics, Takeda, Infotech Soft, Celgene, Merck and has received research support from Janssen Pharmaceuticals and Juno Therapeutics, and declares ownership in CellSpace Biosciences. P.D.G is on the Scientific Advisory Board of Celsius, Earli, Elpiscience, Immunoscape, Rapt, and Nextech, was a scientific founder of Juno Therapeutics, and receives research support from Lonza. J.D.G. declared contracted research with Gilead, Lilly, and Regeneron. The remaining authors declare no competing interests.
Figures




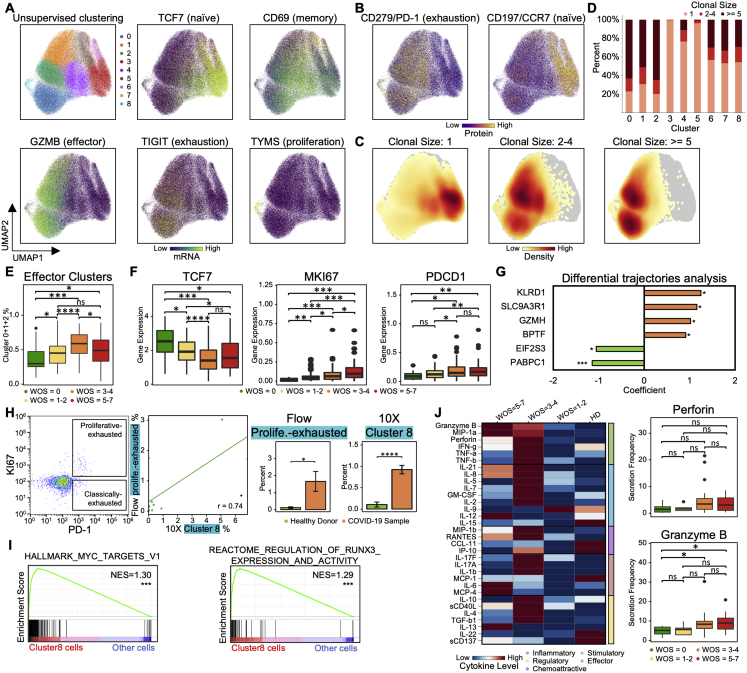
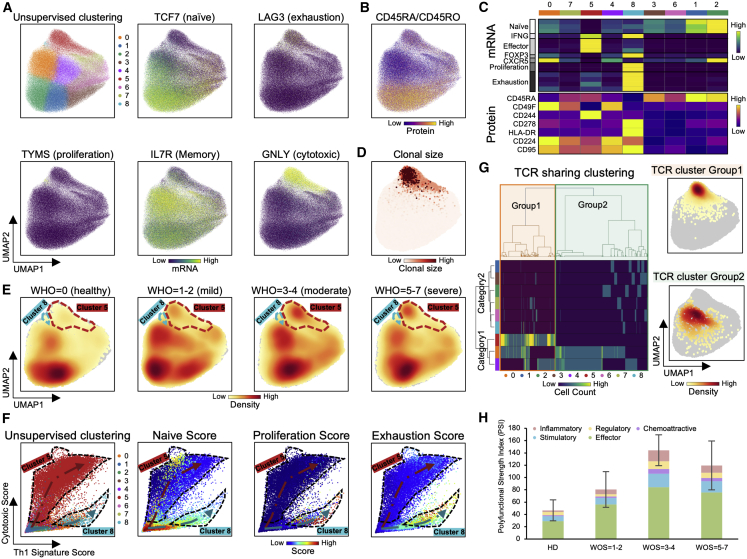
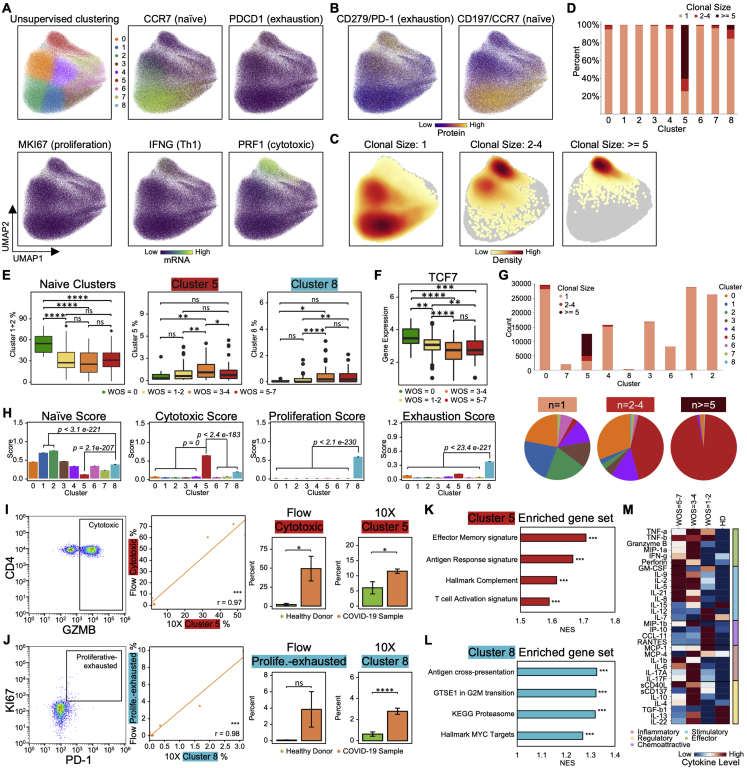
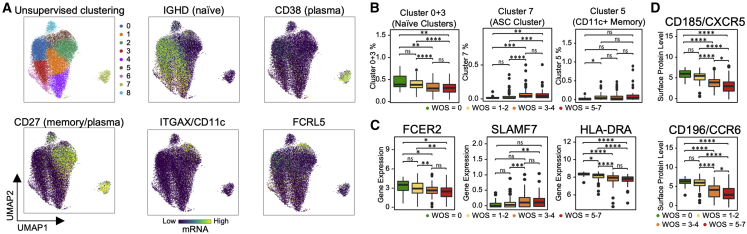

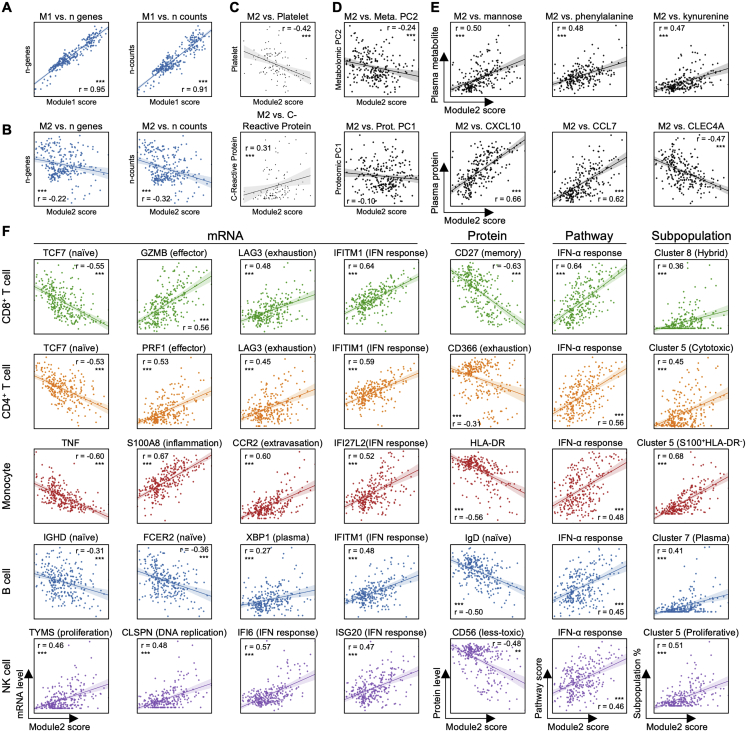
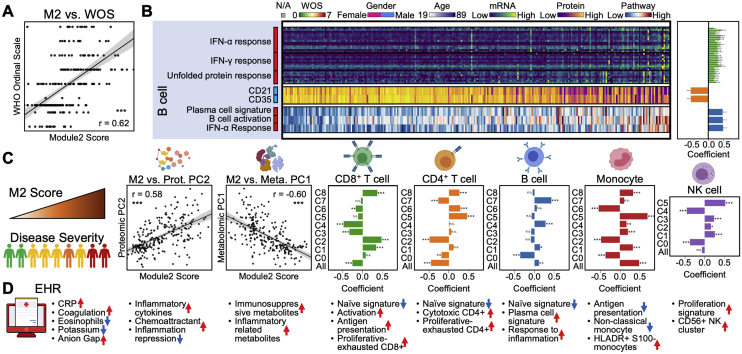
References
-
- Abel B., Tameris M., Mansoor N., Gelderbloem S., Hughes J., Abrahams D., Makhethe L., Erasmus M., de Kock M., van der Merwe L. The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4+ and CD8+ T cells in adults. Am. J. Respir. Crit. Care Med. 2010;181:1407–1417. - PMC - PubMed
-
- Abitorabi M.A., Mackay C.R., Jerome E.H., Osorio O., Butcher E.C., Erle D.J. Differential expression of homing molecules on recirculating lymphocytes from sheep gut, peripheral, and lung lymph. J. Immunol. 1996;156:3111–3117. - PubMed
-
- Agmon N., Alhassid Y., Levine R.D. An algorithm for finding the distribution of maximal entropy. J. Comp. Physiol. 1979;30:250–258.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
