

Research advances in molecular mechanisms underlying the pathogenesis of cystic fibrosis: From technical improvement to clinical applications (Review)
- PMID: 33173976
- PMCID: PMC7646950
- DOI: 10.3892/mmr.2020.11607
Research advances in molecular mechanisms underlying the pathogenesis of cystic fibrosis: From technical improvement to clinical applications (Review)
Abstract
Cystic fibrosis (CF) is a chronic disease causing severe impairment to the respiratory system and digestive tracts. Currently, CF is incurable. As an autosomal recessive disorder, the morbidity of CF is significantly higher among Caucasians of European descent, whereas it is less pervasive among African and Asian populations. The disease is caused by identical mutations (homozygosity) or different mutations (heterozygosity) of an autosomal recessive mutation at position 7q31.2‑q31.1 of chromosome 7. Diagnostic criteria and guidelines work concurrently with laboratory detection to facilitate precise CF detection. With technological advances, the understanding of CF pathogenesis has reached an unprecedented level, allowing for increasingly precise carrier screening, more effective early stage CF intervention and improved prognostic outcomes. These advances significantly increase the life quality and expectancy of patients with CF. Given the numerous improvements in the field of CF, the current review summarized the technical advances in the study of the molecular mechanisms underlying CF, as well as how these improvements facilitate the clinical outcomes of CF. Furthermore, challenges and obstacles to overcome are discussed.
Figures
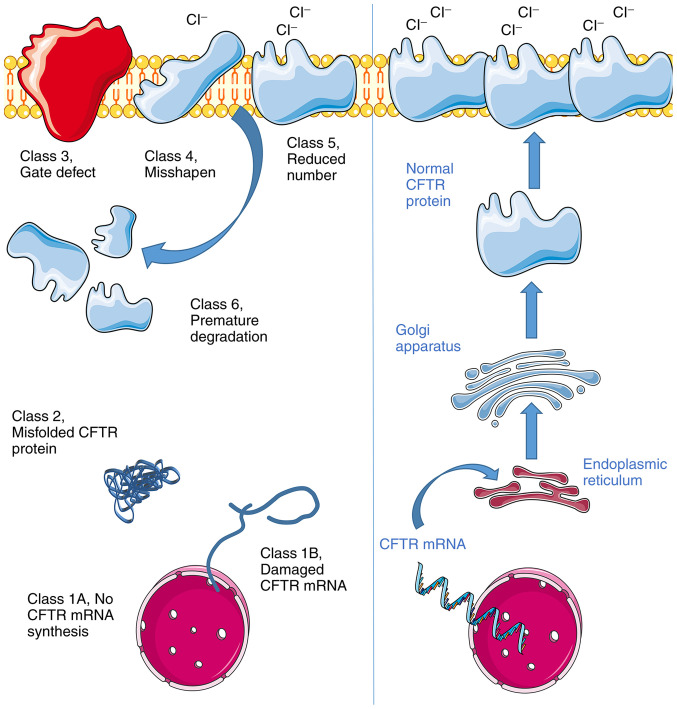
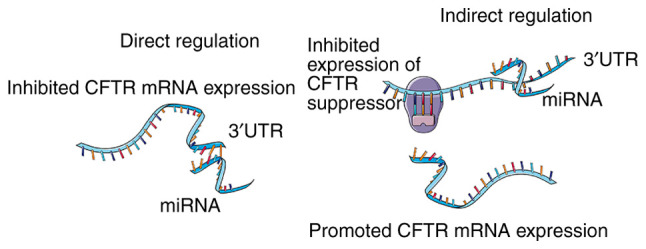
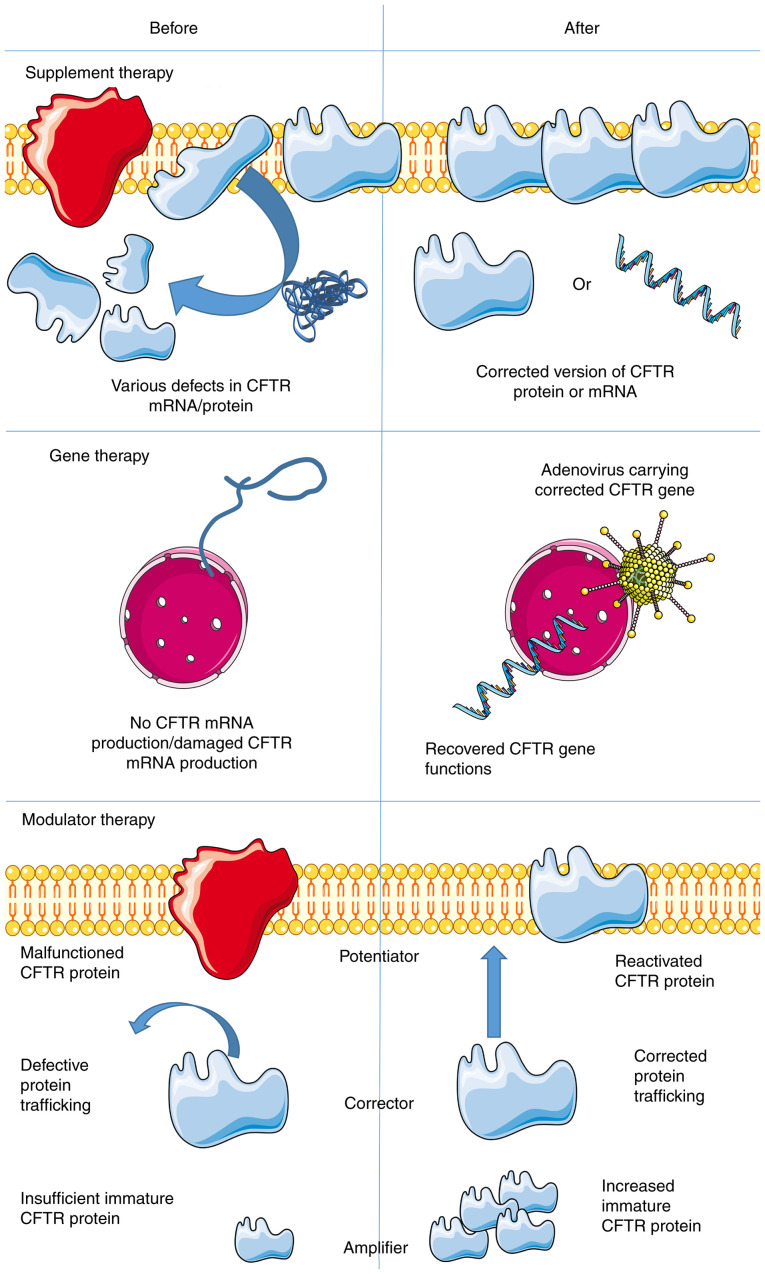
References
-
- Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, Zhou W, Nellis JR, Stroebele EK, Chu KK, et al. Defective innate immunity and hyperinflammation in newborn cystic fibrosis transmembrane conductance regulator-knockout ferret lungs. Am J Respir Cell Mol Biol. 2015;52:683–694. doi: 10.1165/rcmb.2014-0250OC. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
